VERÓNICA GONZÁLEZ-CALLE, RAFAEL FONSECA
Clinical Hematology and Medical Oncology, Mayo Clinic, Scottsdale, USA
Resumen Los estudios de secuenciación masiva realizados recientemente en un gran número de pacientes con mieloma múltiple han permitido profundizar el conocimiento genómico de la enfermedad. La identificación de mutaciones driver como potenciales dianas terapéuticas ofrece una oportunidad para explorar estrategias de tratamiento dirigido a nivel molecular en el mieloma. Por ello, nos encontramos a las puertas de la medicina personalizada, cuyo objetivo fundamental es administrar el tratamiento adecuado a cada paciente según las características concretas de su enfermedad. Este cambio de paradigma es prometedor, aunque a la par surgen nuevos desafíos. A lo largo de esta revisión se describirán los retos fundamentales a afrontar al aplicar la medicina personalizada en mieloma. Además se mencionarán los resultados más relevantes de estudios tanto preclínicos como clínicos con terapias dirigidas en mieloma. Finalmente, se destacará la necesidad de llevar a cabo estudios prospectivos aleatorizados con el fin de evaluar la eficacia de las nuevas terapias dirigidas, así como validar biomarcadores de respuesta que permitan seleccionar los candidatos idóneos para recibir dichos tratamientos, todo ello con el fin de mejorar la supervivencia y calidad de vida de los pacientes con mieloma.
Palabras clave: medicina personalizada, mieloma múltiple, mutación driver, terapia dirigida
Abstract Towards precision medicine in myeloma: new evidence and challenges. In the last few years, next-generation sequencing studies have provided insights into the mutational landscape of multiple myeloma. The identification of actionable mutations might give a precious opportunity for exploring new targeted therapies. Thus, the implementation of promising precision medicine strategies seems to be closer than ever. Throughout this review we describe the main challenges that should to be dealt with in this new era, in order to achieve the main goal of precision medicine, namely matching patients with their right drug. In addition, we provide a review of the most significant preclinical and clinical studies supporting the implementation of precision medicine nowadays. Finally, we highlight the need of clinical trials to evaluate the security and efficacy of these targeted therapies, as well as to validate predictive biomarkers that may allow an appropriate best-candidate selection and improvement of myeloma patients’ survival and quality of life.
Key words: personalized medicine, multiple myeloma, driver mutation, targeted therapy
Recibido: 28-III-2017 Aceptado: 4-V-2017
Dirección postal: Dr. Rafael Fonseca, Mayo Clinic, 5777 East Mayo Boulevard, Phoenix, AZ, EE.UU.
e-mail: fonseca.rafael@mayo.edu
El mieloma múltiple (MM) es una enfermedad neoplásica caracterizada por la proliferación clonal de células plasmáticas en la médula ósea (≥ 10%), la detección de una inmunoglobulina monoclonal en suero u orina y la presencia de anemia, lesiones óseas, hipercalcemia o insuficiencia renal.
En las últimas dos décadas se ha producido una mejora progresiva de la supervivencia de los pacientes con MM, atribuible al desarrollo e incorporación de fármacos muy eficaces como los inhibidores de proteasoma o los inmunomoduladores en la práctica clínica1. Este progreso ha ido por delante del completo conocimiento de la biología de la enfermedad e incluso del mecanismo de acción de estos fármacos. La célula plasmática tumoral comparte características fundamentales con su contrapartida: la célula plasmática normal, productora de inmunoglobulinas, en la que es clave el sistema de ubiquitinización y degradación de proteínas para su homeostasis y supervivencia.
El MM sigue siendo incurable, con períodos de remisión cada vez más cortos seguidos de recaídas más agresivas. En este escenario, resulta prometedor integrar los recientes avances en el conocimiento genómico de la enfermedad, el desarrollo de fármacos dirigidos contra dianas moleculares y trasladarlos a la clínica, desarrollando estrategias de tratamiento personalizadas, con el fin último de lograr un beneficio directo sobre los pacientes, aumentando su supervivencia o incluso su curación.
En esta revisión se describirán algunos de los hallazgos más relevantes proporcionados por los recientes estudios de secuenciación del genoma en el MM y las terapias dirigidas que resultan más prometedoras según dichos resultados.
El camino hacia la medicina personalizada en el MM
El MM es una enfermedad muy heterogénea desde el punto de vista clínico y genético. En los últimos años se han llevado a cabo estudios que han identificado una gran variedad de factores que permiten predecir el curso clínico de los pacientes con MM de forma más aproximada. Algunos como la albúmina sérica o la β2-microglobulina se han integrado en modelos de estratificación de riesgo ampliamente validados como el International Staging System (ISS), o el reciente ISS revisado (R-ISS)2, que incorpora no solo variables relacionadas con el paciente, sino también con la biología de la célula plasmática tumoral, como son las alteraciones citogenéticas de mal pronóstico detectadas por hibridación in situ fluorescente (t(4;14), t(14;16) o del17p) y los niveles de lactato deshidrogenasa. Esta mejor clasificación pronóstica de los pacientes ha permitido evaluar la eficacia de fármacos en subgrupos de pacientes, así como establecer estrategias adaptadas al riesgo en la práctica clínica, como la propuesta por la Clínica Mayo, mSMART, que asegura un tratamiento más intensivo en los pacientes de alto riesgo3 (Tabla 1).
Con la revolución de la genómica y el creciente desarrollo de terapias dirigidas hacia dianas moleculares, nos encontramos en un tiempo de transición hacia la medicina personalizada en cáncer. Uno de los retos actuales es la identificación y validación de biomarcadores moleculares específicos que permitan predecir la respuesta a terapias dirigidas. Sirva de paradigma la leucemia mieloide crónica; donde el cromosoma Philadelphia, t(9;22) o el oncogén BCR/ABL, permiten el diagnóstico, un tratamiento dirigido con inhibidores de tirosina quinasa (que bloquean selectivamente la proteína de fusión BCR-ABL controlando la enfermedad) y una monitorización de la respuesta mediante la detección del producto de fusión por técnicas de citogenética o biología molecular.
El desarrollo de una medicina de precisión en el MM, en particular, afronta varios retos (Tabla 2). En primer lugar, el MM es una enfermedad genéticamente muy heterogénea como se discutirá a continuación. Clásicamente, los MM se han subdividido en hiperdiploides, con ganancias cromosómicas, y no hiperdiploides caracterizados por la presencia de traslocaciones que afectan al gen de la cadena pesada de las inmunoglobulinas (IGH). Los recientes estudios de secuenciación masiva en paralelo del genoma o exoma completo de casi 1000 pacientes con MM han confirmado una gran heterogeneidad y complejidad también a nivel genómico4-7. Además han permitido profundizar el conocimiento de aspectos moleculares claves como el perfil mutacional y los patrones de evolución clonal. A diferencia de otras neoplasias hematológicas como la tricoleucemia con mutaciones en BRAF o la macroglobulinemia de Waldenström en MYD88, en el MM no se ha encontrado una alteración molecular única y específica. Se han identificado varios genes que están recurrentemente mutados: KRAS, NRAS, FAM46C, DISC3 and TP53, aunque casi la mitad de los pacientes presenta mutaciones en otros genes no recurrentes4-6. Por tanto, el diseño de fármacos dirigidos hacia una alteración única en el MM no parece una estrategia terapéutica que vaya a permitir la curación de todos los pacientes.
Otra consideración importante es la heterogeneidad clonal presente en el MM. La mayoría de los genes

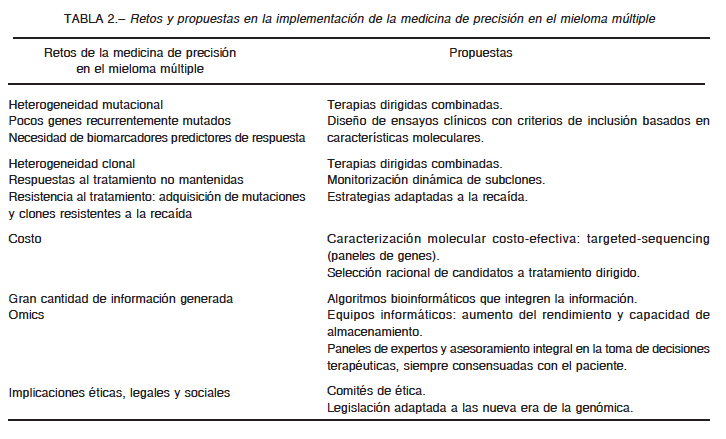
mutados lo están a frecuencias intermedias o bajas, es decir las mutaciones no están presentes en todas las células plasmáticas tumorales, sino en una fracción o subclón tumoral6, 8. En este sentido, se podría especular que un fármaco dirigido contra una alteración molecular específica presente solo en una proporción de células tumorales afectará únicamente a ese subclón, siendo resistentes otros posibles subclones, y por tanto, con un beneficio clínico incompleto. Por otro lado, se debe tener en cuenta la presencia de mutaciones concomitantes en genes pertenecientes a una misma vía de señalización6, pues puede implicar resistencia a terapias dirigidas contra esa vía en particular. En vista de todo lo anterior, las estrategias de tratamiento personalizadas basadas en un único fármaco parecen una aproximación poco realista, y serán necesarias combinaciones según las alteraciones moleculares detectadas en cada paciente.
Por otro lado, la administración continuada de una terapia dirigida ejerce una presión selectiva sobre la población de células tumorales y su microambiente, que puede modificar la evolución de las mismas y facilitar la selección de aquellos subclones con una ventaja adaptativa. De hecho, la administración de tratamientos dirigidos se ha asociado a respuestas cortas, precisamente por la aparición de subclones resistentes que han adquirido una mutación en dominios de unión del fármaco a la proteína diana. Por tanto, la implementación de estas estrategias se deberá acompañar de una monitorización dinámica de los distintos subclones, con el fin de administrar combinaciones de fármacos en el contexto clínico y biológico adecuado.
Terapias dirigidas en desarrollo
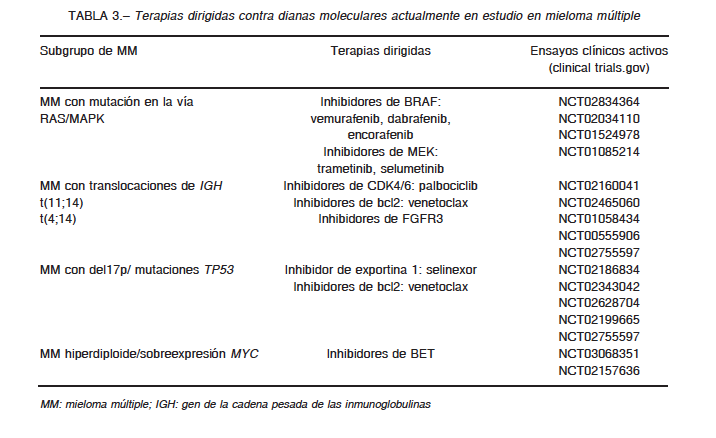
A continuación se revisaran algunas de las dianas moleculares más relevantes en distintos subgrupos de pacientes con MM (Tabla 3).
Mieloma múltiple con mutaciones driver en la vía RAS/MAPK (RAF-MEK-ERK)
Según los estudios de secuenciación masiva en paralelo recientemente publicados, las vías de señalización que con mayor frecuencia presentan mutaciones en genes driver en MM son: RAS/MAPK (40%), NFkB (20%) y vías de reparación de ADN (TP53, ATM) (15%)4-7. La primera de ellas, resulta interesante por ser la más frecuentemente alterada y por la disponibilidad de terapias dirigidas que ya se han investigado en otros tipos de cáncer. En 4% de los pacientes con MM de nuevo diagnóstico se encuentran mutaciones en BRAF V600E. Los inhibidores selectivos de BRAF han mostrando actividad antitumoral en MM tanto in vitro como in vivo. Las series de casos publicadas muestran que el inhibidor selectivo de BRAF vemurafenib es eficaz en pacientes con MM refractario con mutaciones en BRAF 9, constituyendo una buena evidencia para explorar estos agentes en MM en el contexto de ensayos clínicos. No obstante, uno de los principales problemas es la breve duración de la respuesta y el riesgo de desarrollar tumores secundarios (carcinoma escamoso y leucemia) debidos a la hiperactivación de ERK. En este sentido la inhibición conjunta con inhibidores de MEK como trametinib y de BRAF podría reducir dicho riesgo y aumentar la eficacia.
Actualmente hay ensayos clínicos en marcha evaluando la seguridad y eficacia de estos fármacos (Tabla 3).
Mieloma múltiple con translocaciones de IGH
La desregulación de ciclinas es un evento común a los distintos tipos de MM, con un papel clave en la patogénesis de la enfermedad que no se ha descifrado completamente. En 15-20% de los MM está presente la t (11;14) que asocia sobreexpresión del gen de la ciclina D1 por yuxtaposición del enhancer de IGH. En el momento actual los inhibidores de quinasas dependientes de ciclina (CDK4/6) como palbociclib o abemaciclib están siendo evaluados en distintos tipos de cáncer. Resultados preliminares de estudios tanto preclínicos como clínicos con palbociclib muestran actividad antitumoral en MM10.
Además, los pacientes con t(11;14) tratados con venetoclax, un inhibidor selectivo de la proteína antiapoptótica Bcl-2, en ensayos clínicos fase I-II11 presentaron una tasa de respuesta mayor que otros subgrupos de pacientes con MM. Actualmente se está llevando a cabo un estudio fase III (NCT02755597) para determinar la eficacia de este nuevo fármaco en combinación con el estándar (bortezomib y dexametasona). Según estudios preliminares, la presencia de t(11;14), niveles elevados de Bcl-2 y bajos de Mcl-1 o Bcl-XL, así como la presencia de marcadores de célula B podrían predecir la respuesta a venetoclax y permitirían identificar los candidatos idóneos para recibir dicho fármaco. Aún son necesarios estudios prospectivos que validen estos biomarcadores12, 13.
En 15% de pacientes con MM se detecta la traslocación t(4;14), asociada a mal pronóstico. Este subgrupo de pacientes se beneficia de tratamiento con inhibidores de proteasoma como bortezomib14. Además se han explorado otras posibles dianas terapéuticas como FGRFR3 ya que este receptor tirosina quinasa está sobre-expresado en los pacientes con t(4;14) y puede ser inhibido selectivamente con inhibidores de tirosina quinasa. En el momento actual se están llevando a cabo estudios clínicos para evaluar la eficacia y seguridad de estos fármacos en MM.
Mieloma múltiple con del17p o mutaciones en TP53
La deleción del17p está presente en aproximadamente 10% de los MM de nuevo diagnóstico, siendo esta proporción mayor en la recaída, asociándose a enfermedad extramedular y peor supervivencia. Es más, según los resultados del Myeloma XI trial, únicamente las mutaciones en genes de la vía de reparación de ADN, como TP53 o ATM, se asocian a mal pronóstico7
Los fármacos aprobados en la actualidad para el tratamiento del MM no han logrado revertir el mal pronóstico relacionado con la presencia de esta alteración. Desde el punto de vista biológico, la célula plasmática tumoral con pérdida de p53 no responde adecuadamente al daño en el ADN producido por la quimioterapia convencional, por lo que no se induce la muerte celular mediada por p53. De ahí que sea necesario el desarrollo de fármacos con mecanismos de acción distintos. En el momento actual hay dos estrategias trasladadas a la clínica que resultan interesantes en este sentido. En primer lugar, los inhibidores de exportina 1(XPO-1), como selinexor, evitan que proteínas como p53 salgan del núcleo al citoplasma a través de la XPO-1, permaneciendo así activas y funcionales como supresores tumorales. Según los resultados preliminares del ensayo clínico STORM15, selinexor podría estabilizar la
enfermedad en los pacientes con del17p. Por otro lado, estudios preclínicos han demostrado que venetoclax promueve la apoptosis de forma independiente a p53. Además, recientemente se ha publicado el caso de un paciente con MM refractario con t(11;14) y del17p con una respuesta mantenida bajo tratamiento con venetoclax y dexametasona16. Otra estrategia terapéutica que aún no ha sido evaluada en clínica pero que resulta muy interesante en pacientes con mutaciones en TP53 es PRIMA-1. Esta molécula interacciona con p53 mutada y le devuelve su conformación wild-type restaurando su actividad como supresor tumoral y llevando a la célula a la apoptosis17.
Mieloma múltiple hiperdiploide y
sobreexpresión de MYC
La mitad de los MM que se diagnostican son hiperdiploides. Muchos de estos asocian sobreexpresión de MYC, con un impacto negativo sobre el pronóstico de los pacientes, de ahí que sea interesante explorar estrategias para atacar esta diana. Una de las más prometedoras actúa sobre mecanismos epigenéticos: mediante inhibidores de BET se impide el reconocimiento de histonas acetiladas y, en consecuencia, el inicio de la transcripción en genes como MYC. Estudios in vitro han demostrado que el inhibidor de BET JQ1 tiene efecto antiproliferativo en MM, aunque por el momento no hay disponibles resultados de estudios clínicos18.
En resumen, aunque en el momento actual no hay evidencia suficiente para implementar la medicina de precisión en el MM en la práctica clínica, la revolución genómica y el desarrollo de fármacos dirigidos contra dianas moleculares permiten vislumbrar los albores de lo que será una prometedora era. El diseño de nuevos ensayos clínicos basados en la presencia de alteraciones moleculares concretas podría ayudar en la identificación de biomarcadores de respuesta y evaluación de la eficacia de las terapias dirigidas. Por el momento, debemos aguardar los resultados de los ensayos clínicos en marcha evaluando los fármacos mencionados en esta revisión. Otros grandes desafíos como la gran heterogeneidad genómica harán necesaria la administración de terapias combinadas con el fin de obtener respuestas sostenidas, así como la implementación de estrategias de seguimiento que permitan adoptar tratamientos basados en la dinámica de la evolución clonal.
Agradecimientos: La Dra. Verónica González-Calle tiene una beca Fundación Española de Hematología y Hemoterapia-Janssen (Beca Estancias de Investigación en el Extranjero Convocatoria 2015-2016).
Conflicto de intereses: El Dr. Rafael Fonseca es consultor de AMGEN, BMS, Celgene, Takeda, Bayer, Jansen, Novartis, Pharmacyclics, Sanofi y Merck. Miembro del Scientific Advisory Board de Adaptive Biotechnologies. La Mayo Clinic y el Dr. Fonseca tienen una patente de pronóstico mediante FISH con ingresos anuales de unos US$ 2000.
Bibliografía
1. Kumar SK, Rajkumar SV, Dispenzieri A, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood 2008;111: 2516-20.
2. Palumbo A, Avet-Loiseau H, Oliva S, et al. Revised International Staging System for Multiple Myeloma: a report from International Myeloma Working Group. J Clin Oncol 2015; 33: 2863-9.
3. Mikhael JR, Dingli D, Roy V, et al. Management of newly diagnosed symptomatic multiple myeloma: updated Mayo Stratification of Myeloma and Risk-Adapted Therapy (mSMART) consensus guidelines 2013. Mayo Clin Proc 2013; 88: 360-76.
4. Chapman MA, Lawrence MS, Keats JJ, et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011; 471: 467-72.
5. Bolli N, Avet-Loiseau H, Wedge DC, et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun 2014; 5: 2997.
6. Lohr JG, Stojanov P, Carter SL, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell 2014; 25: 91-101.
7. Walker BA, Boyle EM, Wardell CP, et al. Mutational spectrum, copy number changes, and outcome: results of a sequencing study of patients with newly diagnosed myeloma. J Clin Oncol 2015; 33: 3911-20.
8. Kortuem KM, Braggio E, Bruins L, et al. Panel sequencing for clinically oriented variant screening and copy number detection in 142 untreated multiple myeloma patients. Blood Cancer J 2016; 6: e397.
9. Andrulis M, Lehners N, Capper D, et al. Targeting the BRAF V600E mutation in multiple myeloma. Cancer Discov 2013; 3: 862-9.
10. Niesvizky R, Badros AZ, Costa LJ, et al. Phase 1/2 study of cyclin-dependent kinase (CDK)4/6 inhibitor palbociclib (PD-0332991) with bortezomib and dexamethasone in relapsed/refractory multiple myeloma. Leuk Lymphoma 2015; 56: 3320-8.
11. Kumar S, Vij R, Kaufman JL, et al. Venetoclax monotherapy for relapsed/refractory multiple myeloma: safety and efficacy results from a Phase I study. Blood 2016; 128:488.
12. Gupta VA, Newman S, Bahlis NJ, et al. B-cell markers predict response to venetoclax in multiple myeloma. Blood 2016; 128: 2108.
13. Wu J, Ross J, Peale FV, et al. A favorable BCL-2 family expression profile may explain the increased susceptibility of the t(11;14) multiple myeloma subgroup to single agent venetoclax. Blood 2016; 128: 5613.
14. Avet-Loiseau H, Leleu X, Roussel M, et al. Bortezomib plus dexamethasone induction improves outcome of patients with t(4;14) myeloma but not outcome of patients with del(17p). J Clin Oncol 2010; 28: 4630-4.
15. Vogl DT, Dingli D, Cornell RF, et al. Selinexor and low dose dexamethasone (Sd) in patients with lenalidomide, pomalidomide, bortezomib, carfilzomib and anti-CD38 Ab refractory multiple myeloma (MM): STORM Study. Blood 2016; 128: 491.
16. Touzeau C, Le Gouill S, Mahé B, et al. Deep and sustained response after venetoclax therapy in a patient with very advanced refractory myeloma with translocation t(11;14). Haematologica 2017;102: e112-e4.
17. Teoh PJ, Bi C, Sintosebastian C, et al. PRIMA-1 targets the vulnerability of multiple myeloma of deregulated protein homeostasis through the perturbation of ER stress via p73 demethylation. Oncotarget 2016; 7: 61806-19.
18. Affer M, Chesi M, Chen WD, et al. Promiscuous MYC locus rearrangements hijack enhancers but mostly super-enhancers to dysregulate MYC expression in multiple myeloma. Leukemia 2014; 28: 1725-35.