
TATIANA POTES1, SANTIAGO GALICCHIO2, BÁRBARA ROSSO3, GABRIELA BESOCKE3, MARÍA DEL CARMEN GARCÍA3, JUAN CARLOS AVALOS3
1Servicio de Neurología, Hospital Italiano Buenos Aires, 2Sanatorio de Niños, Rosario, Santa Fe, 3Sección Epilepsia, Servicio de Neurología, Hospital Italiano Buenos Aires, Argentina
Resumen La enfermedad de Lafora es infrecuente; sin embargo, es una de las causas más comunes de epilepsia mioclónica progresiva. Presentamos el caso de una mujer de 19 años sin comorbilidades y pautas madurativas normales, que inició a los 8 años con convulsiones y que a partir de los 15 años agregó deterioro cognitivo progresivo. Fue internada en nuestra institución con diagnóstico de estatus epiléptico super refractario. Se diagnosticó enfermedad de Lafora, confirmada por la anatomía patológica, y posteriormente se realizó un test genético que informó una variante patogénica del gen EPM2A, que confirmó el diagnóstico. Presentamos una causa de epilepsia mioclónica progresiva, con un pronóstico ominoso y un tratamiento orientado a medidas paliativas, por lo que es importante analizar los diagnósticos diferenciales con otras entidades, a fin de establecer un pronóstico, ofrecer mejor calidad de vida, asistencia médica adecuada y brindar asesoría genética a los familiares.
Palabras clave: estatus epiléptico, epilepsia mioclónica progresiva, enfermedad de Lafora
Abstract Progressive myoclonic epilepsy secondary to Lafora’s body disease. Lafora’s disease is infrequent. However, it is one of the most common causes of progressive myoclonus epilepsy. We present the case of a 19-year-old woman, without comorbidities and normal development that started at 8 years with seizures and that from 15 years, had progressive cognitive deterioration. She was admitted to our institution with a diagnosis of super refractory status epilepticus. The diagnosis of Lafora’s disease was made through pathological anatomy, later a genetic test was performed that reported a pathogenic variant of the EPM2A gene, confirming the diagnosis. We present a cause of progressive myoclonic epilepsy, with an ominous prognosis and a treatment oriented to palliative measures, so it is important to analyze the differential diagnoses with other entities, in order to establish a prognosis, offer better quality of life, adequate medical care and provide genetic counseling to family members.
Key words: status epilepticus, progressive myoclonic epilepsy, Lafora’s disease
Recibido: 11-VI-2018 Aceptado: 18-VII-2018
Dirección postal: Juan Carlos Avalos, Sección Epilepsia, Servicio de Neurología, Hospital Italiano Buenos Aires, Tte. Gral. Juan Domingo Perón 4230, 1190 Buenos Aires, Argentina
e-mail: juancavalos.neurologia@gmail.com
La enfermedad de Lafora (EL) es una epilepsia mioclónica autosómica recesiva. Es particularmente frecuente en países del Mediterráneo (España, Francia, Italia), norte de África y en algunas regiones de India1. La EL comienza clásicamente en la adolescencia en individuos neurológicamente normales, y los primeros síntomas son crisis mioclónicas y tónico-clónicas, crisis de ausencia, atónicas o convulsiones focales de inicio en el área visual. Pueden también presentarse síntomas neuropsiquiátricos. Comúnmente, en las fases iniciales de la enfermedad puede confundirse con una epilepsia mioclónica juvenil; sin embargo, presenta una progresión rápida que involucra trastornos mnésicos, epilepsia refractaria, psicosis, ataxia, disartria, apraxia, ceguera y muerte en aproximadamente una década2. La EL es causada por mutaciones en los genes EPM2A o EPM2B (NHLRC1), que codifican la laforina fosfatasa de doble especificidad y la malin ubiquitina E3 ligasa respectivamente, involucradas en el metabolismo del glucógeno3, 4.
Al inicio de la enfermedad, la resonancia de cerebro (RM) no muestra cambios, pero con el tiempo puede mostrar atrofia cortical. Las anomalías en el electroencefalograma (EEG) a menudo preceden a los síntomas clínicos y pueden observarse lentificación o actividad paroxística, focal o generalizada. Los depósitos de poliglucanos en diferentes tejidos como glándulas sudoríparas, miocitos, hepatocitos e incluso el cerebro, son fundamentales para su aproximación diagnóstica.
Caso clínico
Mujer de 19 años, sin comorbilidades y pautas madurativas normales. A los 8 años de edad inició con convulsiones de variada semiología, incluidas focales con pérdida de conciencia, mioclónicas y tónico-clónicas generalizadas, por lo que recibió diferentes esquemas terapéuticos (lamotrigina, fenitoína, perampanel, carbamazepina, vigabatrina, topiramato, etosuximida y dieta cetogénica) sin respuesta al tratamiento médico. A los 15 años de edad presentó deterioro cognitivo progresivo que la obligó a discontinuar sus estudios escolares. Dado el aumento en la frecuencia de convulsiones fue internada para ajuste del tratamiento farmacológico antiepiléptico e intercurrió con estado de mal epiléptico. No respondió a la primera y segunda línea de tratamiento, por lo cual recibió drogas anestésicas. Tras el descenso de la medicación anestésica presentó nuevamente convulsiones, por lo cual fue derivada a nuestra institución con diagnóstico de estado de mal epiléptico super refractario. Ingresó a la unidad de terapia intensiva en sedoanalgesia con propofol, midazolam, remifentanilo y drogas antiepilépticas (levetiracetam y lacosamida). La RM de cerebro evidenció atrofia cortical difusa, sin lesiones focales. El monitoreo videoelectroencefalográfico mostró la presencia de actividad theta rítmica con actividad rápida y espigas superpuestas en región fronto-central bilateral, en coincidencia con artificio muscular correspondiente a mioclónica palpebrales y presencia de actividad epileptiforme interictal en la región témporo-parieto-occipital bilateral. Dados estos hallazgos, se progresó con el tratamiento farmacológico hasta obtener registro eléctrico de paroxismo supresión, pero frente al descenso de la medicación instaurada se observaba nuevamente actividad epileptiforme continua. En razón de su estado de mal epiléptico super refractario, se buscó alternativas farmacológicas que incluyeron tratamiento inmunológico con inmunoglobulinas, hipotermia, estimulación magnética transcraneal y ketamina. Con la sospecha diagnóstica de epilepsia mioclónica progresiva, se realizó biopsia de piel donde se observó inclusiones intracitoplasmáticas PAS (ácido peryódico reactivo de Schiff) positivas en acinos glandulares (Fig. 1). Para su confirmación diagnóstica se realizó un test genético que informó una variante patogénica del gen EPM2A, confirmándose el diagnóstico de enfermedad de Lafora. En cuanto a la evolución, se produjo el deceso durante su internación en unidad de terapia intensiva6, 7.
Discusión
La enfermedad de Lafora o síndrome de Lafora fue descripta por primera vez en 1911 por el neuropatólogo español Gonzalo Rodríguez Lafora. La epilepsia mioclónica progresiva autosómica recesiva tiene una frecuencia de 1-4 casos / 1 000 000 y un inicio en personas jóvenes con desarrollo previo normal. Los primeros síntomas de la enfermedad incluyen convulsiones del tipo mioclónicas, tónico-clónico generalizadas, de ausencia, atónicas y convulsiones de inicio focal con manifestaciones visuales. La disminución en el rendimiento escolar, depresión y apatía son también de aparición temprana en la enfermedad5. Estos antecedentes fueron constatados en la paciente, lo cual nos permitió su sospecha diagnóstica. Comúnmente, en los años posteriores los síntomas progresan con convulsiones refractarias al tratamiento médico, psicosis, ataxia y demencia. La muerte usualmente ocurre dentro de los 10 años, a menudo por neumonía por aspiración asociada a estado de mal epiléptico. Tanto el inicio clínico, como la evolución y desenlace final, coinciden con lo descrito en la literatura.
Las anomalías del EEG pueden preceder a los síntomas clínicos, inicialmente consisten en un fondo lento, actividad generalizada o focal con predominio en la región occipital, no acentuada por el sueño, siendo la mioclonía y la marcada fotosensibilidad características destacadas6, 7.
La EL se caracteriza por la presencia de inclusiones intracelulares PAS positivas, que consisten en un depósito anormal de glucógeno llamado poliglicanos, que se acumulan en hígado, corazón, piel, músculos y neuronas. Este hallazgo fue encontrado en nuestra paciente, pero su interpretación puede estar sujeta a falsos positivos, dado que requiere experiencia para distinguir los polisacáridos normales de las glándulas sudoríparas apocrinas8. Por lo tanto, recurrimos a la confirmación genética, que en este
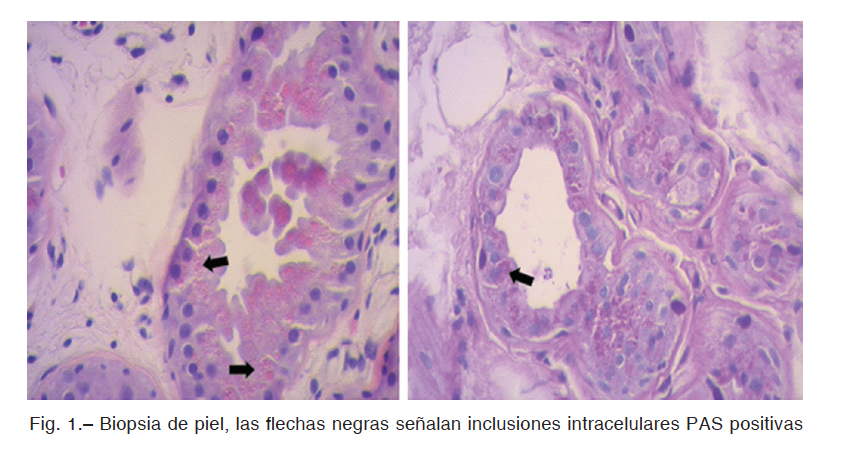
caso consistió en una alteración en el gen EMP2A. La EL es causada por mutaciones en el gen EPM2A o EPM2B que codifican la proteína fosfatasa laforina y la ubiquitina respectivamente, involucradas en el metabolismo del glucógeno9.
Las correlaciones genotipo-fenotipo no revelan diferencias sustanciales entre los pacientes, pero algunas mutaciones específicas del EPM2B pueden correlacionarse con un inicio tardío y lentamente progresivo10.
La EL forma parte de un grupo de enfermedades englobadas en el término epilepsia mioclónica progresiva (EMP), entre las cuales se encuentran la enfermedad de Unverricht-Lundborg, la lipofuscinosis neuronal ceroidea, la epilepsia mioclónica con fibras rojas rasgadas y la sialidosis. Las EMP son trastornos neurodegenerativos hereditarios caracterizados por epilepsia mioclónica, disfunción neurológica variable y empeoramiento progresivo (Tabla 1).
La EL es la principal causa de EMP en adolescentes. La enfermedad de Unverricht-Lundborg es causada por mutaciones en el gen de la cistatina B, y es el diagnóstico diferencial más frecuente de la EL. Sin embargo, en la enfermedad de Unverricht-Lundborg, las mioclonías de acción y las convulsiones tónico-clónicas son más controlables y el deterioro cognitivo generalmente es leve.
En la lipofuscinosis se ha encontrado alteraciones en al menos nueve genes y la mayoría codifican proteínas lisosomales. Estos pacientes exhiben acumulación de lipofuscina en diferentes órganos y, en su mayoría, los síntomas se presentan en la primera infancia. La forma juvenil de la lipofuscinosis (enfermedad de Batten, gen CLN3) aparece más tardíamente que la enfermedad de Lafora, e inicia con un periodo prolongado de pérdida visual y convulsiones leves. Puede haber una presentación infantil de la lipofuscinosis (enfermedad de Santavuori- Haltia; CLN1) y una forma muy rara de presentación adulta (enfermedad de Kufs). La epilepsia mioclónica con fibras rojas rasgadas es una mitocondriopatía que muestra niveles elevados de lactato en sangre y en líquido cefalorraquídeo, junto con fibras rojas irregulares en la biopsia de músculo. La sialidosis en una rara enfermedad
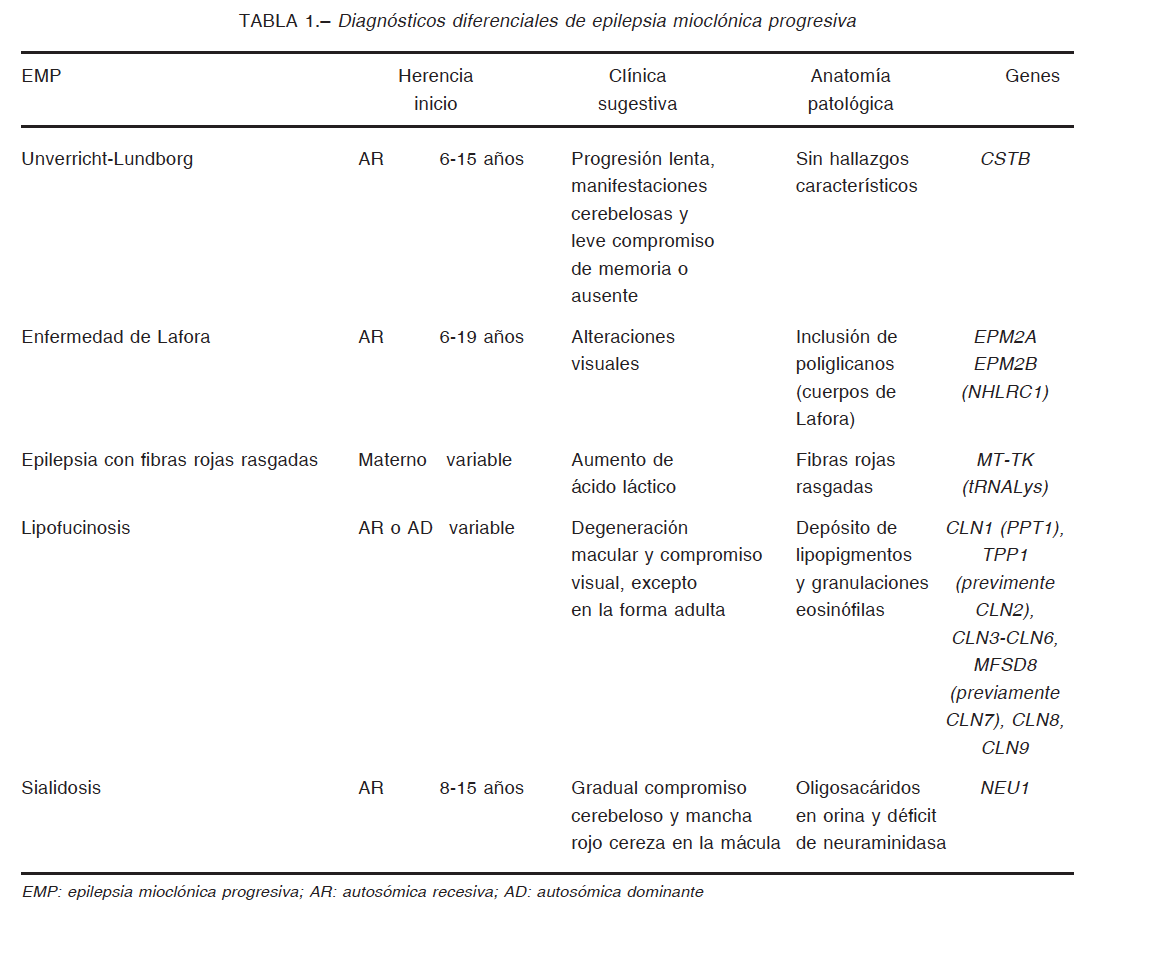
lisosomal asociada a maculopatía con mancha rojo cereza y niveles elevados de oligosacáridos en orina.
Presentamos un caso de estado de mal epiléptico superrefractario, en cuyo contexto se realizó diagnóstico de enfermedad de Lafora con confirmación genética. Esta es una de las causas de epilepsia mioclónica progresiva más frecuente, aunque con un pronóstico ominoso y un tratamiento orientado a medidas paliativas, por lo que es importante analizar los diagnósticos diferenciales con otras EMP y procurar su confirmación, a fin de establecer un pronóstico, ofrecer mejor calidad de vida, asistencia médica adecuada y brindar asesoría genética a los familiares.
Conflicto de intereses: Ninguno para declarar
Bibliografía
1. Minassian BA. Lafora’s disease: towards a clinical, pathologic, and molecular synthesis. Pediatr Neurol 2001; 25: 21-9.
2. Striano P, Zara F, Turnbull J, et al. Typical progression of myoclonic epilepsy of the Lafora type: a case report. Nat Clin Pract Neurol 2008; 4: 106-11.
3. Chan EM, Bulman DE, Paterson AD, et al. Genetic mapping of a new Lafora progressive myoclonus epilepsy locus (EPM2B) on 6p22. J Med Genet 2003; 40: 671-5.
4. Chan EM, Young EJ, Ianzano L, et al. Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat Genet 2003; 35: 125-7.
5. Ohi H, Chan EM, Scherer SW, Minassian BA. On the road to tractability: the current biochemical understanding of progressive myoclonus epilepsies. Adv Neurol 2006; 97: 399-415.
6. Boccella P, Striano P, Zara F, et al. Bioptically demonstrated Lafora disease without EPM2A mutation: a clinical and neurophysiological study of two sisters. Clin Neurol Neurosurg 2003; 106: 55-9.
7. Canafoglia L, Ciano C, Panzica F, et al. Sensorimotor cortex excitability in Unverricht-Lundborg disease and Lafora body disease. Neurology 2004; 63: 2309-15.
8. Andrade DM, Ackerley CA, Minett TS, et al. Skin biopsy in Lafora disease: genotype-phenotype correlations and diagnostic pitfalls. Neurology 2003; 61: 1611-4.
9. Gentry MS, Worby CA, Dixon JE. Insights into Lafora disease: malin is an E3 ubiquitin ligase that ubiquitinates and promotes the degradation of laforin. Proc Natl Acad Sci USA 2005; 102: 8501-6.
10. Lohi H, Turnbull J, Zhao XC, et al. Genetic diagnosis in Lafora disease: genotype-phenotype correlations and diagnostic pitfalls. Neurology 2007; 68: 996-1001.
– – – –
Como lo habrá observado el lector, la lucha entre la sociedad y sus instituciones es el rasgo predominante del sistema. Un conjunto de sentimientos, el culto nacional del coraje, el desprecio de la ley, la preocupación exclusiva de la fortuna, la fe en la grandeza del país, imprimen rumbos fijos a la sociedad. El derecho político argentino comienza a formarse. Se ve su esquema confusamente trazado, con los caracteres esenciales que conservará siempre, no obstante los nombres exóticos y la literatura constitucional yanqui: predominio del concepto clásico de Estado-providencia, centralización política, papel inferior y subordinado de las asambleas; y en el pueblo, para acentuar y fortificar esas tendencias, el desprecio de la ley convertido en instinto, en uno de los motivos de la voluntad.
Juan Agustín García (1862-1923)
La ciudad indiana (1900). Buenos Aires; Emecé, s/f; p 299