
DANIEL NATERA DE BENITO 1, CARLOS ORTEZ 1, LAURA CARRERA GARCÍA 1, JESSICA EXPÓSITO 1, EDNA BOBADILLA 1, ANDRÉS NASCIMENTO 1, 2
1 Unidad de Enfermedades Neuromusculares, Servicio de Neurología, Hospital Sant Joan de Déu, Barcelona, 2 Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), Instituto de Salud Carlos III, Madrid, España
Resumen Existen importantes avances en el campo de las miopatías congénitas en los últimos años que obligan a la revisión y actualización constante de este grupo de enfermedades. La identificación creciente de nuevos genes y fenotipos asociados a genes ya conocidos, fue posible en gran medida gracias al avance de las técnicas de secuenciación de nueva generación, cada vez más accesibles. El conocer mejor el espectro fenotípico de estas entidades, permite establecer una correlación fenotipo/genotipo en algunos subgrupos. La mejor compresión de la fisiopatología e historia natural de estas enfermedades, son fundamentales para el desarrollo de nuevas terapias. Los primeros ensayos clínicos en el campo de la terapia génica ya son una realidad y están mostrando resultados positivos, creando una nueva expectativa en paciente, familiares y especialistas, lo que se verá reflejado en la necesidad de adaptar los protocolos de atención, diagnóstico y tratamiento de algunas de estas entidades. Es fundamental que los neuropediatras, pediatras, fisioterapeutas y otros profesionales involucrados en el cuidado de estos pacientes, estén informados y actualizados de los avances en este grupo de enfermedades.
Palabras clave: miopatía congénita, hipotonía, debilidad, genética
Abstract Diagnosis and treatment of congenital myopaties. Important advances have been made in the field of congenital myopathies in recent years, forcing clinicians to constantly review and update this group of diseases. The increasing identification of new genes and phenotypes associated with already known genes has been possible to a great extent thanks to the development accomplished in next generation sequencing techniques, which are increasingly accessible. Knowing better the phenotypic spectrum of these entities allows to establish a phenotype/genotype correlation in some subgroups. The best understanding of the pathophysiology and natural history of these diseases are fundamental to design new therapies. The first clinical trials in the field of gene therapy are already a reality and are showing positive results, creating a new expectation for patients, families and specialists, which will be reflected in the need to adapt the protocols of care, diagnosis and treatment of some of these entities. It is essential that pediatric neurologists, pediatricians, physiotherapists and other professionals involved in the care of these patients are informed and updated on the advances in this group of diseases.
Key words: congenital myopathy, hypotonia, weakness, genetics
Barcelona, España
e-mail: anascimento@sjdhospitalbarcelona.org
El avance de los últimos años en las técnicas utilizadas para el estudio molecular, la comprensión de la fisiopatología y la terapia génica están cambiando el diagnóstico y la terapéutica de las enfermedades neuromusculares 1, 2. Las miopatías congénitas (MC) son un grupo de enfermedades clínica y genéticamente heterogéneo que afecta de forma primaria a la fibra muscular, en especial al aparato contráctil y a los diferentes componentes que condicionan su normal funcionamiento. La mayoría se presenta en forma de debilidad muscular e hipotonía al nacimiento o durante el primer año de vida 1-3.
Características clínicas generales y diagnóstico
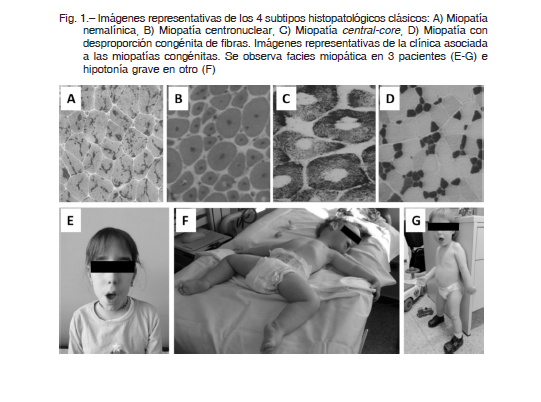
El espectro clínico es amplio, con una gran variabilidad que va desde cuadros de debut neonatal con afectación grave, artrogriposis, dificultad respiratoria, disfagia, hasta cuadros de presentación en la infancia o en el adulto con síntomas leves 3, 4. La distribución de la debilidad suele ser generalizada o más pronunciada en los músculos axiales, proximales y faciales (Fig. 1 E,F,G). El curso de la debilidad suele ser lentamente progresivo o relativamente estable y los niveles de las enzimas musculares son a menudo normales o ligeramente elevados. En general no hay afectación cognitiva. La presencia de cardiomiopatía y el compromiso respiratorio pueden estar presentes en algunos casos y son datos útiles en la orientación diagnóstica 3. La mortalidad en los primeros años de vida es elevada en las formas graves 5, 6. Se han publicado diversos estudios sobre la historia natural 6-9.
La prevalencia global de las miopatías congénitas no se ha determinado con precisión, aunque es probable que ocurra en al menos en 1/20 000 niños 5-9.
El diagnóstico se fundamenta en la clínica y en los hallazgos observados en la anatomía patológica, que
permiten orientar el diagnóstico genético 1, 3. La caracterización de los diferentes patrones evidenciados en la resonancia magnética muscular ha permitido incluir esta técnica como una herramienta útil en el diagnóstico 10. Es importante destacar que es frecuente la superposición de fenotipos histopatológicos y clínicos, lo que a menudo dificulta el diagnóstico y la correlación genotipo/fenotipo 1-3.
El trabajo coordinado e integrado entre clínicos, radiólogos, genetistas, biólogos, bioinformáticos y patólogos con experiencia es la clave para el diagnóstico 1-3.
Clasificación de las miopatías congénitas
El término miopatía congénita fue utilizado por primera vez en 1956 por Shy y Magee en la descripción de un grupo de enfermedades neuromusculares de aparición congénita que comparten características histopatológicas y ultraestructurales en la biopsia muscular, sin mostrar cambios distróficos evidentes 1-4, 11.
Históricamente las MC se han clasificado en cuatro grupos según la característica predominante en la biopsia muscular 1-4, 11-13 (Fig. 1 A, B, C, D y 2 A):
1) Miopatía nemalínica (MN): grupo amplio con agregados protéicos.
2) Miopatía centronuclear (MCN): alteración de la localización del núcleo.
3) Miopatía con cores (MCC): desestructuración del patrón intermiofibrilar.
4) Miopatía por desproporción congénita del tipo de fibras (MDCF)
En los últimos años se han identificado una gran variedad de genes que causan diferentes formas de MC. A pesar de estos avances y el uso cada vez más estandarizado de técnicas de secuenciación masiva, al día de hoy no es posible identificar la mutación o el gen responsable en un 40-50% de los pacientes 12-14.
Se han descrito todas las formas de herencia en el conjunto de las MC. Cabe resaltar que diferentes mutaciones en el mismo gen pueden causar enfermedades musculares muy distintas y mutaciones en diferentes genes pueden causar fenotipos muy similares (Fig. 2 A y B) 3, 12-13.
El avance en el conocimiento de las bases genéticas de las MC está condicionando un cambio en la clasificación nosológica clásica. Un número creciente de genes asociados a las MC que se describen cada año se puede consultar en www.musclegenetable.fr (Gene Table of Neuromuscular Disorders). La clasificación es cada vez más compleja, ya que se debe tener en cuenta el genotipo, fenotipo, histotipo y fisiotipo 2, 12-14. Además, debido a la mejor comprensión de la interrelación de estos pilares, el establecimiento del diagnóstico genético correcto es importante para la atención clínica proactiva-multidisciplinar y las posibles consideraciones terapéuticas. A continuación describiremos las principales características de los cuatro grandes subgrupos de MC.

Miopatías nemalínicas
Se caracterizan por la presencia de cuerpos o bastones nemalínicos en la biopsia muscular (Fig. 1A). Se visualizan mediante tinción de tricrómico de Gomori o mediante microscopía electrónica 4, 11, 12. Dan lugar a un espectro clínico amplio, con presentaciones clínicas desde el periodo neonatal hasta la edad adulta. Como características clínicas comunes encontramos debilidad facial y de la musculatura bulbar, que puede afectar a la deglución y al habla. No suele haber afectación de musculatura extraocular 3, 4. Pueden clasificarse en base al inicio y gravedad de los síntomas, además del compromiso respiratorio en: a) forma congénita grave, b) forma congénita clásica y c) forma del adulto.
Miopatías centronucleares
Este grupo ha sido clásicamente definido por la presencia de núcleos centrales en más del 25% de las fibras musculares (Fig. 1B) 4, 11, 12. Originalmente, esta entidad fue denominada “miopatia miotubular” debido a la similitud con los miotúbulos fetales que se observan en el músculo en desarrollo de fetos sanos. Actualmente, el término miopatía miotubular queda reservado para los casos producidos por mutaciones en el gen MTM1. Otros genes, como DNM2 y BIN1, pueden dar lugar a esta forma de miopatía 12, 13. Clínicamente es heterogénea, con formas de presentaciones desde el período neonatal hasta la edad adulta. La presencia de oftalmoparesia es relativamente común y puede ser útil para el enfoque diagnóstico 3.
Miopatías con cores
Bajo esta denominación queda englobado un grupo clínicamente heterogéneo de miopatías que tienen en común
la presencia en la biopsia muscular de unas estructuras denominadas cores 4, 11, 12. Los cores se definen como áreas circunscritas en las que existe una marcada reducción o una ausencia de la tinción para enzimas oxidativas (Fig. 1 C). Según la morfología del defecto, se dividen en miopatías con central-core (presencia de cores únicos localizados en el centro de la fibra muscular) y miopatías con multiminicore (presencia difusa de numerosos cores de pequeño tamaño) 4, 11.
Las miopatías con cores son el subtipo más frecuente y la mayoría son causadas por mutaciones en los genes RYR1 y SEPN1.
Las mutaciones en SEPN1 dan lugar a un fenotipo reconocible, típicamente caracterizado por una miopatía
con multiminicores, debilidad cérvico-axial y posterior aparición de espina rígida con escoliosis. Los pacientes a menudo mantienen la capacidad de deambular hasta la edad adulta. Es frecuente la asociación de insuficiencia respiratoria precoz y progresiva con necesidad de ventilación asistida 3, 13.
Las mutaciones en RYR1 pueden dar lugar a una MC con cores centrales o con multiminicores. Los pacientes
con central-cores por mutación en RYR1 suelen presentar una debilidad relativamente leve y no progresiva de predominio en extremidades. En cualquier caso, existe una minoría con MC por mutaciones dominantes en RYR1 y central-core que presentan una debilidad grave desde el nacimiento, asociando una discapacidad también grave. La MC por RYR1 y multiminicores suelen estar asociadas a un patrón de herencia autosómico recesivo y una debilidad predominantemente axial, con escoliosis, displasia de caderas, deformidades de la caja torácica y retracciones articulares 3. La presencia de oftalmoparesia es frecuente en las miopatías por RYR1 recesivo y no en las miopatías por RYR1 dominante. Las mutaciones en RYR1 pueden dar lugar a cualquiera de los 4 subtipos histológicos de MC. Algunos pacientes pueden asociar susceptibilidad a hipertermia maligna (desencadenada por el empleo de algunos anestésicos volátiles y relajantes musculares) 4, 11-14.
Existen otros genes que también pueden dar lugar a una miopatía con central-core: MYH7 (típicamente
asociado a miopatía distal de Laing), ACTA1, KBTBD13, CCDC78 y TTN 13.
Miopatías por desproporción congénita de fibras
Las manifestaciones clínicas en este subgrupo están más relacionadas con el gen alterado que con los hallazgos histopatológicos. Los genes que se relacionan más con este grupo de miopatías son TPM3, TPM2, RYR1 y ACTA 4, 11-13.
Para hacer el diagnóstico de MC por desproporción del tamaño de las fibras debe existir 1) clínica de miopatía y 2) en la biopsia deben observarse unas fibras tipo 1 al menos un 35-40% más pequeñas que las de tipo 2 (Fig. 1D) 11, 12.
Los pacientes con este tipo de miopatía presentan hipotonía neonatal y debilidad generalizada que puede
ser desde leve a muy grave, con debut entre el período neonatal y los primeros años de vida. Existe un retraso en la adquisición de los hitos del desarrollo motor y una debilidad muscular que afecta particularmente a la cintura y a la cara, con una reconocible facies miopática y paladar ojival. Existe disfagia asociada en aproximadamente el 30% de los pacientes, que puede persistir más allá de los primeros meses de vida y requerir gastrostomía 3, 4.
Tratamiento
La mayoría de las MC, con excepción de algunos tipos de comienzo muy precoz y clínica muy grave, tienen un curso clínico y un pronóstico relativamente estable 3,4,15,16.
Actualmente no existe un tratamiento curativo para ninguna de las formas de MC. Por ello, es necesario establecer un plan específico de seguimiento para cada paciente que garantice la mejor calidad de vida posible y que incluya cuidados de rehabilitación, cardíacos, respiratorios y deglutorios 16. Es recomendable alcanzar un diagnóstico genético, puesto que la causa genética de la enfermedad tiene implicaciones a nivel de pronóstico y de prevención.
Por ejemplo, un seguimiento respiratorio estrecho es especialmente recomendable en pacientes con mutaciones en los genes NEB, MTM1, ACTA1 y SEPN1; mientras que el seguimiento cardíaco es especialmente importante en pacientes con mutaciones en MYH7 y TPM2 1, 3, 4, 15, 16. Una capacidad vital forzada en la espirometría menor del 60% sobre el valor predicho para la edad, es de utilidad clínica para sospechar la presencia de una hiperventilación nocturna 1, 3, 4, 16. La miocardiopatía se asocia excepcionalmente a la MC, pero debe seguirse estrechamente cuando existen mutaciones en determinados genes como TTN y MYH7 1, 4. En cualquiera de los subtipos se recomienda llevar a cabo controles cardiológicos periódicos, que incluirán tanto ecocardiograma como electrocardiograma 15, 16.
Desde el punto de vista ortopédico debe ofrecerse una atención individualizada que incluya tratamientos conservadores o quirúrgicos de las retracciones articulares y de la escoliosis. La fisioterapia para mantener una adecuada función articular es útil, así como un ejercicio aeróbico regular. Existe un riesgo aumentado en estos pacientes de osteoporosis y fracturas, por lo que se recomienda un control de la densidad ósea y valoración de la indicación de suplementos de calcio y vitamina D 1, 3, 4, 15, 16.
Diferentes estudios han demostrado la afectación de la unión neuromuscular en algunos pacientes con MC.
Existen comunicaciones de casos con buena respuesta a los anticolinestarásicos y al salbutamol 17, 18. No debemos olvidar la importancia de ofrecer métodos de comunicación aumentativo, terapia ocupacional y logopedia desde las primeras etapas 15, 16. De la misma forma, el apoyo psicológico al paciente y su familia es necesario y de utilidad para ayudar a integrar los diferentes aspectos vinculados con la asimilación del diagnóstico, la reformulación de expectativas familiares y dar ideas sobre cómo explicarle al niño su enfermedad, sus capacidades y limitaciones de manera que se facilite un buen afrontamiento y adaptación a la discapacidad física tanto en el entorno familiar como escolar 16.
Recientemente se han publicado resultados positivos de la terapia génica en modelos animales de miopatía
miotubular, lo que ha sentado las bases para el desarrollo de ensayos en pacientes 19. ASPIRO fase 1/2 es un ensayo clínico que está en curso para evaluar la seguridad y la eficacia preliminar. Los sujetos reciben una dosis única de AT132 y son seguidos por seguridad y eficacia durante 5 años (NCT03199469). Un análisis interino presentado en el Congreso Mundial de la Sociedad del Músculo de 2018 informó un aumento en la fuerza de extremidades y tronco, mejoría en la movilidad global, capacidad de comunicación, manejo de secreciones y capacidad de deglución 20. A la espera de que la llegada de estos tratamientos pueda modificar la historia natural de la enfermedad, tiene una enorme importancia ofrecer a los pacientes un seguimiento multidisciplinario óptimo para potenciar su calidad de vida e integración 1, 4, 15, 16.
Agradecimientos: Agradecemos a los pacientes con miopatías congénitas y sus familiares, al Servicio de Anatomía Patológica y al equipo del Biobanco del Hospital Sant Joan de Déu por la colaboración en el estudio de muestras de músculo y compartir la imágenes realizadas de las biopsias musculares utilizadas en este artículo.
Conflicto de intereses: Ninguno para declarar
Bibliografía
1. Gonorazky HD, Bönnemann CG, Dowling JJ. The genetics of congenital myopathies. Handb Clin Neurol 2018; 148: 549-64.
2. Vita G, Vita GL, Musumeci O, Rodolico C, Messina S. Genetic neuromuscular disorders: living the era of a therapeutic revolution. Part 2: diseases of motorneuron and skeletal muscle. Neurol Sci 2019; 40: 671-81.
3. Gonorazky HD, Dowling JJ, Volpatti JR, Vajsar J. Signs and symptoms in congenital myopathies. Semin Pediatr Neurol 2019; 29: 3-11.
4. Schorling DC, Kirschner J, Bönnemann CG. Congenital muscular dystrophies and myopathies: an overview and update. Neuropediatrics 2017; 48: 247-61.
5. Annoussamy M, Lilien C, Gidaro T, et al. X-linked myotubular myopathy: A prospective international natural history study. Neurology 2019; 92: e1852- 67.
6. Fox MD, Carson VJ, Feng HZ, et al. TNNT1 nemaline myopathy: natural history and therapeutic frontier. Hum Mol Genet 2018; 27: 3272-82.
7. Beggs AH, Byrne BJ, De Chastonay S, et al. A multicenter, retrospective medical record review of X-linked myotubular myopathy: The recensus study. Muscle Nerve 2018; 57: 550-60.
8. Amburgey K, Tsuchiya E, de Chastonay S, et al. A natural history study of X-linked myotubular myopathy. Neurology 2017; 89:1355-64.
9. Colombo I, Scoto M, Manzur AY, et al. Congenital myopathies: Natural history of a large pediatric cohort. Neurology 2015; 84: 28-35.
10. Carlier RY, Quijano-Roy S. Myoimaging in congenital myopathies. Semin Pediatr Neurol 2019; 29: 30-43.
11. North KN, Wang CH, Clarke N, et al. Approach to the diagnosis of congenital myopathies. Neuromuscul Disord 2014; 24: 97-116.
12. Phadke R. Myopathology of congenital myopathies: bridging the old and the new. Semin Pediatr Neurol 2019; 29: 55-70.
13. Pelin K, Wallgren-Pettersson C. Update on the genetics of congenital myopathies. Semin Pediatr Neurol 2019; 29: 12-22.
14. Radke J, Stenzel W, Goebel HH. Recently identified congenital myopathies. Semin Pediatr Neurol 2019; 29: 83-90.
15. Jungbluth H, Muntoni F. Therapeutic aspects in congenital myopathies. Semin Pediatr Neurol 2019; 29: 71-82.
16. Wang CH, Dowling JJ, North K, et al. Consensus statement on standard of care for congenital myopathies. J Child Neurol 2012; 27: 363-82.
17. Illingworth MA, Main M, Pitt M, et al. RYR1-related congenital myopathy with fatigable weakness, responding to pyridostigimine. Neuromuscul Disord 2014; 24: 707-12.
18. Rodríguez Cruz PM, Sewry C, Beeson D, et al. Congenital myopathies with secondary neuromuscular transmission defects; a case report and review of the literature. Neuromuscul Disord 2014; 24: 1103-10
19. Elverman M, Goddard MA, Mack D, et al. Long-term effects of systemic gene therapy in a canine model of myotubular myopathy. Muscle Nerve 2017; 56: 943-53.
20. Kuntz N, Shieh P, Smith B, et al. ASPIRO phase 1/2 gene therapy trial in X-linked myotubular myopathy: preliminary safety and efficacy findings. Neuromuscul Disord 2018; 28 (Suppl 2): S91.