
LUCIANO E. MARASCO, JOSÉ STIGLIANO, ALBERTO R. KORNBLIHTT
Instituto de Fisiología, Biología Molecular y Neurociencias (IFIBYNE), UBA-CONICET, Departamento de Fisiología, Biología Molecular y Celular, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Argentina
Resumen El splicing alternativo del ácido ribonucleico mensajero (mRNA) juega un papel fundamental en el flujo de información genética desde el ADN a las proteínas al expandir la capacidad de codificación de los genomas. La regulación del splicing alternativo es tan importante como la regulación de la transcripción para determinar las características específicas de las células y los tejidos, el funcionamiento normal de las células y las respuestas de las células eucarióticas a las señales externas. El conocimiento básico de las secuencias del pre-mRNA y de los factores de splicing que las reconocen ha permitido a científicos diseñar un oligonucleótido sintético terapéutico para la atrofia muscular espinal. Ésta es una enfermedad hereditaria autosómica recesiva en que el gen SMN1 se encuentra mutado y que afecta a uno de cada 10 000 nacimientos. Al bloquear la unión de un factor de splicing negativo al mRNA del gen parálogo del gen SMN1, denominado SMN2, el oligonucleótido Spinraza corrige un evento de splicing alternativo anormal del gen SMN2 y permite que se sinteticen altos niveles de la proteína SMN, constituyéndose en el primer caso exitoso de cura de una enfermedad neurode generativa.
Palabras clave: splicing alternativo, atrofia muscular espinal, enfermedades hereditarias
Abstract Alternative splicing of the messenger RNA plays a fundamental role in the flow of genetic information from DNA to proteins by expanding the coding capacity of the genome. The regulation of alternative splicing is as important as the regulation of transcription to determine the specific characteristics of cells and tissues, the normal functioning of cells and the responses of eukaryotic cells to external signals. Basic knowledge of the pre-mRNA sequences and splicing factors that recognize them has allowed scientists to design a therapeutic synthetic oligonucleotide for spinal muscular atrophy. This is an autosomal recessive inherited disease in which the SMN1 gene is mutated and affects one in 10,000 births. By blocking the binding of a negative splicing factor to the mRNA of a paralogue of the SMN1 gene, called SMN2, the Spinraza oligonucleotide corrects an abnormal alternative splicing event of the SMN2 gene and allows the synthesis of high levels of the SMN protein, constituting the first successful case of cure of a neurodegenerative disease.
Key words: alternative splicing, spinal muscular atrophy, hereditary disease
Dirección postal: Dr. Alberto R. Kornblihtt, IFIBYNE (UBA-CONICET), Facultad de Ciencias Exactas y Naturales, Ciudad Universitaria, Pabellón IFIBYNE, 1428 Buenos Aires, Argentina
e-mail: ark@fbmc.fcen.uba.ar
Cada uno de los genes que codifican proteínas en los eucariotas pluricelulares tiene regiones que estarán representadas en el ácido ribonucleico (RNA) mensajero (mRNA) maduro, intercaladas por otras cuyas secuencias que no estarán representadas allí. Las primeras regiones se llaman exones, en tanto que las segundas son los intrones. La enzima que transcribe el gen, llamada RNA polimerasa II (RNAPII), fabrica un RNA precursor, llamado transcripto primario o pre-RNA mensajero (pre-mRNA), que lleva información tanto de los exones como de los intrones. Dentro del núcleo, y de manera simultánea a la transcripción del gen, un complejo molecular formado por proteínas y RNA llamado “spliceosoma”, elimina los intrones y une los exones entre sí, mediante un mecanismo denominado splicing en inglés, o corte y empalme o ayuste en castellano. Finalmente, el RNA mensajero maduro sin intrones abandona el núcleo y es traducido por los ribosomas del citoplasma celular, que fabrican la proteína correspondiente.
Splicing alternativo
La naturaleza partida de los genes eucarióticos, organizados en exones e intrones, y la aparición concomitante de splicing de pre-mRNA parecen haber tenido al menos dos ventajas evolutivas. A nivel filogenético, podrían generarse nuevos genes a partir de los ancestros a través de una recombinación no disruptiva en los intrones en un proceso conocido como barajado de exones 1. La segunda ventaja evolutiva es el splicing alternativo, un mecanismo que opera a nivel ontogenético al permitir que un solo gen genere dos o más variantes de mRNA maduro, expandiendo la capacidad de codificación de los genomas eucarióticos. El número de genes que codifican proteínas en vertebrados no es muy diferente del número en los invertebrados: alrededor de 20 000 genes tanto en humanos como en el gusano microscópico Caenorhabditis elegans. Sin embargo, el número de genes cuya expresión ocurre mediante splicing alternativo y el número promedio de isoformas de mRNA generadas por gen son mayores en los vertebrados 2, lo que sugiere fuertemente que la prevalencia del splicing alternativo en estos organismos es importante por su mayor complejidad. Por ejemplo, el splicing alternativo ocurre en casi el 95% de los genes de mamíferos 3, 4, pero solo en un 35% de los genes de C. elegans. El splicing alternativo puede ejemplificarse con una metáfora que visualiza al mRNA maduro como un tren formado por numerosos vagones (los exones) enganchados entre sí y donde existen dos formaciones distintas del tren, una con el vagón comedor colocado en el medio y otra sin él. Diríamos que el tren tiene dos formaciones posibles, pues en el caso de los genes decimos que el gen puede originar dos mRNA distintos y, por lo tanto, dos proteínas distintas.
El pre-mRNA es el sustrato de la maquinaria de splicing. Éste se realiza gracias a que las ribonucleoproteínas que componen el “spliceosoma” reconocen secuencias consenso cortas en el pre-mRNA conocidas como sitios de splicing, ubicadas al comienzo y al final de cada intrón. Los sitios de splicing “fuertes” (es decir, aquellos que están más ajustados a la secuencia consenso) se reconocen y utilizan más eficazmente que los sitios de splicing “débiles” o subóptimos. Es la proximidad de sitios de splicing fuertes y débiles que compiten a lo largo de un pre-mRNA naciente que conduce a un splicing alternativo 5, 6.
Enhancers y silenciadores del splicing
El splicing alternativo está regulado tanto por secuencias que actúan en cis como por factores que operan en trans, llamados globalmente factores de splicing. Los primeros incluyen enhancers y silenciadores del splicing exónicos (ESE y ESS respectivamente), y enhancers y silenciadores del splicing intrónicos (ISE y ISS respectivamente). Los factores que actúan en trans funcionan mediante la unión a enhancers y silenciadores del splicing e incluyen miembros de las familias de proteínas SR (proteínas ricas en serina arginina) y hnRNP (heterogeneous nuclear ribonucleoproteins), así como factores específicos de tejidos. Algunos de estos factores activan, mientras que otros inhiben, el uso de sitios de splicing y, en algunos casos, la dirección del efecto depende de la posición del sitio de unión en el pre-mRNA 5.
Acoplamiento del splicing alternativo con la transcripción
El splicing alternativo no solo está regulado por la abundancia y las modificaciones post-traduccionales de los factores de splicing, sino también por las interacciones funcionales y físicas entre las maquinarias de transcripción y de splicing. Hace más de 20 años, nuestro laboratorio encontró que diferentes promotores de RNAPII colocados frente a la misma unidad transcripcional provocan diferentes proporciones de dos isoformas de splicing provenientes del mismo gen 7, 8, lo que introdujo la idea del acoplamiento del splicing alternativo con la transcripción. Posteriormente se demostró que no solo los promotores, sino también los factores de transcripción y los coactivadores, los enhancers de la transcripción, los remodeladores de cromatina y los factores que afectan la estructura de la cromatina influyen en las decisiones del splicing alternativo.
Se han propuesto dos mecanismos, que no son mutuamente excluyentes, para explicar el acoplamiento entre la transcripción y el splicing: acoplamiento por reclutamiento y acoplamiento cinético. El primero implica que la maquinaria de la transcripción recluta factores de splicing que operan sobre el pre-mRNA a medida que éste es fabricado por la RNAPII. En el segundo, variaciones en la velocidad con que la RNAPII recorre el gen, en lo que se conoce como elongación de la transcripción, condicionan el uso de sitios débiles y fuertes del splicing por parte de los componentes del spliceosoma y afectan las proporciones de las isoformas del splicing alternativo.
La prueba más directa del acoplamiento cinético involucró el uso de una mutante “lenta” de la RNAPII. La mutación consiste en la sustitución de un aminoácido en el dominio catalítico de su subunidad mayor, y muestra una tasa de elongación de la transcripción reducida tanto in vitro como in vivo 9, 10. Se demostró que la transcripción llevada a cabo por esta mutante aumenta la inclusión de varios exones alternativos, tanto en estudios de genes individuales 11 como globales en protocolos de secuenciación masiva de mRNA 12.
En la forma más frecuente del acoplamiento cinético, que afecta a aproximadamente el 80% de los exones alternativos sensibles a la velocidad de transcripción, la elongación lenta causa una mayor inclusión del exón alternativo porque favorece el reclutamiento de factores splicing positivos a sitios de splicing débiles que se encuentran río arriba de otros más fuertes antes de que estos últimos sean sintetizados por la RNAPII. En la forma menos frecuente, que afecta al 20% remanente, ocurre el efecto contrario sobre ciertos exones, es decir, la lentitud produce mayor exclusión al crear una ventana de oportunidad para que factores de splicing negativos se unan a sus secuencias “blanco” en el pre-mRNA. Recientemente nuestro grupo describió el mecanismo por el cual la inclusión en el mRNA de un exón alternativo del gen CFTR (cystic fibrosis transmembrane regulator) es menor cuando la velocidad de elongación es más lenta 13. Por lo tanto, los efectos positivos o negativos que puede tener el acoplamiento cinético en la inclusión de exones dependerán de la identidad y la arquitectura de las secuencias reguladoras particulares que rodean cada evento de empalme alternativo.
Cromatina y splicing alternativo
En el núcleo, las moléculas de ADN están asociadas con proteínas llamadas histonas formando estructuras con forma de “tambor” conocidas como nucleosomas. Los nucleosomas forman unidades repetitivas fundamentales de la estructura cromatínica, permitiendo comprimir el voluminoso genoma eucariota dentro del núcleo y a la vez permitir un acceso adecuado al mismo. Los nucleosomas están organizados como una fibra de forma análoga a la estructura de un “collar de perlas”, conectadas por segmentos de ADN desnudo libre de histonas (ADN linker).
En su forma más compacta, conocida como heterocromatina, los nucleosomas se aproximan entre sí y dificultan tanto la unión de proteínas reguladoras de la transcripción de los genes como el avance de la RNAPII. Lo opuesto ocurre cuando los nucleosomas se alejan unos de otros formando eucromatina. En este escenario, el ADN se relaja y la RNAPII avanza más eficientemente.
Nuestro grupo ha demostrado que el grado de compactación de la cromatina en torno a la región que ocupa un exón que sufre splicing alternativo tiene un impacto directo sobre los niveles de su inclusión. Los tratamientos o condiciones que promueven la compactación de la cromatina afectan los niveles de inclusión de exones alternativos específicos dado que la RNAPII puede avanzar más lentamente 14, 15. En general, la metilación de ciertos residuos aminoacídicos de las histonas, como por ejemplo la lisina 9 de la histona H3 (H3K9me2), promueve la compactación de la cromatina. Recíprocamente, los tratamientos o condiciones que relajan la estructura cromatínica, tales como la acetilación del mismo residuo de histona (H3K9Ac) promueven el efecto opuesto 13. Estos efectos dependen del exón alternativo específico involucrado. El laboratorio también demostró que la introducción en células de RNAs pequeños de interferencia (siRNAs) (de 20-25 nucleótidos de largo) que interaccionan con secuencias de intrones cercanos a exones alternativos, modifica el patrón de inclusión de exones en el mRNA maduro a través de inducir cambios en la condensación de la cromatina y en la velocidad de transcripción 16, 17, representando una nueva forma de control del splicing alternativo.
La cura de la atrofia muscular espinal a través del control del splicing alternativo
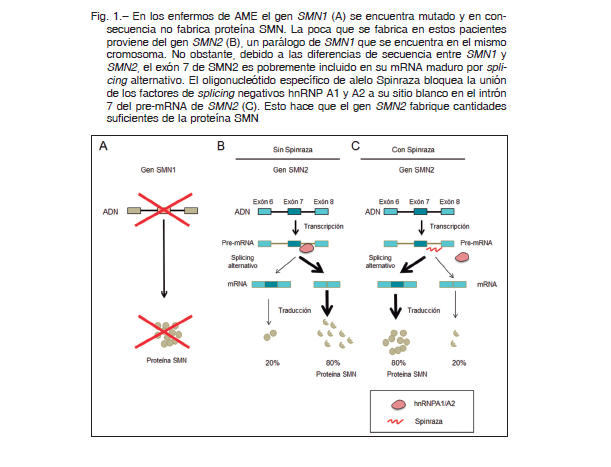
La atrofia muscular espinal (AME en castellano y SMA en inglés, por spinal muscular atrophy) es una grave enfermedad hereditaria, autosómica recesiva, que afecta a uno de cada 10 000 nacidos. La AME es la causa genética más importante de mortalidad infantil. Está causada por mutaciones de pérdida de función en el gen SMN1 (survival motor neuron 1). Los bebés que heredaron las dos copias mutadas del gen SMN1 no producen suficiente cantidad de la proteína SMN (Fig. 1A). Esta proteína es un componente importante de la maquinaria del splicing. Si bien su ausencia debería causar efectos múltiples, las motoneuronas de la médula espinal parecen ser más sensibles que el resto de las células del organismo a la disminución de la cantidad de SMN y su degeneración hace que los pacientes vayan perdiendo la capacidad de contraer sus músculos, tanto los de sus extremidades como los de su caja torácica. Sin asistencia respiratoria mecánica externa, los bebés afectados de la forma más grave de AME (la tipo I) morirían asfixiados a los pocos meses de haber nacido.
Aun así, sin movilidad muscular, los pacientes con AME tipo I quedan postrados de por vida.
En diciembre de 2016, se aprobó un tratamiento que tiene un impresionante poder curativo de la AME. El investigador que encontró la cura es el biólogo molecular uruguayo Adrián Krainer, que trabaja desde hace más de 30 años en los EE.UU., y dirige un laboratorio en Cold Spring Harbor en las afueras de Nueva York. La estrategia de Krainer no consistió en “curar” al gen mutado SMN1 sino en aprovechar que en los humanos existe un gen parálogo, llamado SMN2, que podría funcionar como respaldo, o “backup” de SMN1, porque su producto también es la proteína SMN. El problema es que SMN2 posee 11 cambios de base en su secuencia al compararlo con SMN1. Entre esos cambios, se altera la secuencia óptima de un enhancer del splicing localizado en su exón 7. Esto hace que el exón 7 se incluya pobremente en el mRNA maduro de SMN2 y, por consiguiente, predomine la variante de splicing alternativo no funcional que carece del dicho exón y es por ello que el gen SMN2 no produce suficiente cantidad de proteína SMN para prevenir la neurodegeneración (Fig. 1B). Gracias a su detallado estudio de las secuencias en cis y de los factores en trans que regulan el splicing alternativo, Krainer identificó en el intrón 7 un ISS que une los factores de splicing hnRNP A1 y A2 que actúan negativamente sobre la inclusión del exón 7.
Con este conocimiento, Krainer diseñó un oligonucleótido sintético con una secuencia de bases complementaria a la del ISS del intrón 7. Al introducir este segmento corto de ácido nucleico en las células, el mismo se aparea con su secuencia “blanco” en el pre-mRNA de SMN2 y, por competencia, bloquea la unión de hnRNP A1 y A2 con lo cual logra que el gen SMN2 produzca cantidades suficientes de la proteína SMN, cosa que el gen SMN1 no puede hacer por estar mutado 18 (Fig. 1C). Este tipo de estrategia no está incluida dentro de lo que se conoce como terapia génica, sino que es una terapia por oligonucleótidos antisentido específicos de alelo (ASO, allelespecific oligonucleotides). Después de múltiples ensayos en células en cultivo, ratones 19-21, monos y pacientes humanos, el ASO, cuyo nombre comercial es Spinraza, fue aprobado por la FDA en 2016 y por la ANMAT en 2019.
El oligonucleótido antisentido Spinraza está modificado químicamente con un grupo 2’-O-(2-metoxetil) (MOE) fosforotioato18 que inhibe su degradación en los fluidos corporales y es administrado por inyección intratecal, es decir, en el líquido cefalorraquídeo.
Krainer recibió numerosos premios porque fue el primer científico en curar una enfermedad neurodegenerativa.
El reconocimiento no solo vino del mundo académico sino también, y muy especialmente, de los padres y familiares de los chicos enfermos.
Nuestro laboratorio en Buenos Aires también se ha dedicado a estudiar el control del splicing alternativo desde hace más de 25 años. Nunca habíamos encarado la cura de una enfermedad hasta que hace 4 años, impulsados por los familiares de pacientes argentinos con AME, nucleados en FAME (Familias Atrofia Muscular Espinal Argentina), comenzamos un proyecto de investigación que se plantea utilizar los conocimientos descubiertos en nuestro grupo para generar una terapia combinada que sume nuestras herramientas al ya consagrado tratamiento con Spinraza. Establecimos una colaboración científica con el Dr. Krainer, obtuvimos financiamiento no solo de FAME sino también de CureSMA, la fundación de familiares de pacientes con AME de EE.UU., dos discípulos ganaron becas doctorales del CONICET para llevar a cabo los experimentos, y al día de hoy podemos decir que los resultados obtenidos, por ahora en células cultivadas y en ratones, son muy prometedores. A grandes rasgos, comprobamos que si tratamos a las células con drogas que generan modificaciones post-traduccionales de las histonas que relajan la cromatina, al provocar que la RNAPII transcriba más rápido que lo normal, el efecto de Spinraza en lograr que se fabriquen cantidades adecuadas de la proteína SMN es mucho mayor, con lo cual podría aplicarse una terapia combinada entre Spinraza y drogas que modifican la cromatina, abaratando quizás el costo de Spinraza que ronda, en el ámbito privado, en más de 100 000 dólares por aplicación, debiéndose efectuar 3 o 4 aplicaciones al año.
Ciencia básica y medicina
El trabajo de Krainer y su grupo es uno de los mejores ejemplos de la importancia de la investigación básica para beneficio de la sociedad. Es imposible obtener resultados transferibles a la medicina, la agricultura o la industria sin una fuerte y bien financiada investigación básica. Es imprescindible conocer al detalle los mecanismos de funcionamiento de las células y sus genes para poder encarar nuevas terapias. No hay atajos. Sin el conocimiento de cómo funciona el splicing, habría sido imposible desarrollar una estrategia exitosa para curar una enfermedad tan devastadora como la AME.
Agradecimientos: Agradecemos a FAME (Familias Atrofia Muscular Espinal Argentina) por su apoyo, compromiso y ejemplo.
Conflicto de intereses: Ninguno para declarar
Bibliografía
1. Hynes RO. The evolution of metazoan extracellular matrix. J Cell Biol 2012; 196: 671-9.
2. Keren H, Lev-Maor G, Ast G. Alternative splicing and evolution: diversification, exon definition and function. Nat Rev Genet 2010; 11: 345-55.
3. Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 2008; 40: 1413-5.
4. Barash Y, Calarco JA, Gao W, et al. Deciphering the splicing code. Nature 2010; 465: 53-9.
5. Kornblihtt AR, Schor IE, Allo M, Dujardin G, Petrillo E, Muńoz MJ. Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nat Rev Mol Cell Biol 2013; 14: 153-65.
6. Ast G. How did alternative splicing evolve? Nat Rev Genet 2004; 5: 773-82.
7. Cramer P, Cáceres JF, Cazalla D, et al. Coupling of transcription with alternative splicing: RNA pol II promoters modulate SF2/ASF and 9G8 effects on an exonic splicing enhancer. Mol Cell 1999; 4: 251-8.
8. Cramer P, Pesce CG, Baralle FE, Kornblihtt, AR. Functional association between promoter structure and transcript alternative splicing. Proc Natl Acad Sci USA 1997; 94: 11456-60.
9. Chen Y, Chafin D, Price DH, Greenleaf AL. Drosophila RNA polymerase II mutants that affect transcription elongation. J Biol Chem 1996; 271: 5993-9.
10. Boireau S, Maiuri P, Basyuk E, et al. The transcriptional cycle of HIV-1 in real-time and live cells. J Cell Biol 2007; 179: 291-304.
11. de la Mata M, Alonso CR, Kadener S, et al. A slow RNA polymerase II affects alternative splicing in vivo. Mol Cell 2003; 12: 525-32.
12. Maslon, MM, Braunschweig, U, Aitken, S, et al. A slow transcription rate causes embryonic lethality and perturbs kinetic coupling of neuronal genes. EMBO J 2019; 38: e101244.
13. Dujardin G, Lafaille C, de la Mata M et al. How slow RNA polymerase II elongation favors alternative exon skipping. Mol Cell 2014; 54: 683-90.
14. Schor IE, Rascovan N, Pelisch F, Alló M, Kornblihtt AR. Neuronal cell depolarization induces intragenic chromatin modifications affecting NCAM alternative splicing. Proc Natl Acad Sci USA 2009; 106: 4325-30.
15. Schor IE, Fiszbein A, Petrillo E, Kornblihtt AR. Intragenic epigenetic changes modulate NCAM alternative splicing in neuronal differentiation. EMBO J 2013; 32: 2264-74.
16. Alló M, Buggiano V, Fededa JP, et al. Control of alternative splicing through siRNA-mediated transcriptional gene silencing. Nat Struct Mol Biol 2009; 16: 717-24.
17. Alló M, Agirre E, Bessonov S, et al. Argonaute-1 binds transcriptional enhancers and controls constitutive and alternative splicing in human cells. Proc Natl Acad Sci USA 2014; 111: 15622-9.
18. Hua Y, Vickers TA, Okunola HL, Bennett CF, Krainer AR. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet 2008; 82: 834-48.
19. Hua Y, Sahashi K, Hung G, et al. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev 2010; 24: 1634-44.
20. Hua Y, Sahashi K, Rigo F, et al. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 2011; 478: 123-6.
21. Hua Y, Liu YH, Sahashi K, Rigo F, Bennett CF, Krainer AR. Motor neuron cell-nonautonomous rescue of spinal muscular atrophy phenotypes in mild and severe transgenic mouse models. Genes Dev 2015; 29: 288-97.