
BEATRIZ CALLEJA-PÉREZ 1, ANA L. FERNÁNDEZ-PERRONE 2, DANIEL M. FERNÁNDEZ-MAYORALAS 2, ANA JIMÉNEZ DE DOMINGO 2, PILAR TIRADO 3, SONIA LÓPEZ-ARRIBAS 4, REBECA SUÁREZ-GUINEA 4, ALBERTO FERNÁNDEZ-JAÉN 2, 5
1 Atención Primaria de Pediatría, Centro de Salud Doctor Cirajas, 2 Servicio de Neurología Infantil, Hospital Universitario Quirónsalud, 3 Sección de Neurología Infantil, Hospital Universitario La Paz, Centro CADE, 4 Psiquiatría, Centro CADE, 5 Facultad de Medicina, Universidad Europea de Madrid, Madrid, España
Resumen Los avances en la genética han podido apoyar la sospecha que aportaba la experiencia clínica sobre el gran componente hereditario de la mayor parte de estos trastornos del neurodesarrollo (TND). Los estudios iniciales de heredabilidad, ligamiento o asociación evidenciaron desde los inicios la gran contribución de la variación genotípica a la clínica en general, y a los TND en particular. No debe obviarse la utilidad de los estudios genéticos en el ejercicio clínico, encaminados al diagnóstico etiológico. La mayor parte de los mismos están protocolizados en el estudio de trastornos como la discapacidad intelectual y el autismo; dentro de éstos, la hibridación por arrays cromosómicos ha aportado una mayor rentabilidad diagnóstica respecto a técnicas citogenéticas históricas (3 vs. 10% respectivamente). Sin embargo, la irrupción y rentabilidad de técnicas de genética molecular por secuenciación, particularmente la exómica y genómica en trío, analizando a padres, (tasas diagnósticas del 30-50%), están condicionando la modificación de los algoritmos genéticos en el diagnóstico de trastornos graves del neurodesarrollo. El mayor conocimiento de variantes causales de discapacidad intelectual y autismo está igualmente modificando los modelos teóricos poligénicos establecidos hasta la fecha.
Palabras clave: trastornos del neurodesarrollo, autismo, discapacidad intelectual, genética, arrays, secuenciación
Abstract Advances in genetics have been able to support the clinical suspicion on the large hereditary component of most of these neurodevelopmental disorders (NDD). Initial studies on heritability, linkage or association showed from the beginning the great contribution of genotypic variation to the clinic in general, and to NDD in particular. The effectiveness of genetic studies in clinical practice, targeted to aetiological diagnosis, should not be ignored. Most of these are protocolized in the study of disorders such as intellectual disability and autism; within these, the array comparative genomic hybridization have supported a greater diagnostic effectiveness with respect to historical cytogenetic techniques (3 vs. 10% respectively). However, the irruption and success of molecular genetic sequencing techniques, particularly the exome and genome in trio, analyzing the parents (diagnostic rates of 30-50%), are conditioning the modification of the genetic algorithms in the diagnosis of different NDD. The greater knowledge of causal variants in intellectual disability and autism is also modifying the polygenic theoretical models established to date.
Key words: neurodevelopmental disorders, autism, intellectual disability, genetics, arrays, sequencing
e-mail: aferjaen@telefonica.net
Todos los trastornos del neurodesarrollo (TND) pueden tener un origen ambiental, genético o multifactorial. Desde el punto de vista genético, las evidencias sobre el papel subyacente de numerosos genes en los TND es clara 1-3.
Los antecedentes más importantes para el conocimiento de la carga genética en los TND se han basado históricamente en los estudios de heredabilidad, de ligamiento y de asociación.
Así, la heredabilidad de los TND es realmente elevada, registrándose índices entre 0.5 y 0.9 para diferentes patologías como los trastornos del espectro autista (TEA) 4, la dislexia 5 o el trastorno por déficit de atención/hiperactividad (TDAH) 6.
El mapeo de los loci de riesgo para los TND, a través de estudios de ligamiento, han mostrado numerosas áreas de riesgo, muchas de ellas compartidas entre diferentes TND; alguno de estos loci, se asocian a LODs o valores de ligamiento no paramétrico elevados: 1q23, 3q26.3, 7p14.1, 13q13.3, 15q11, 15q13.3, 19p13 o Xp22.1 para los TEA, 16p13, 17p11, 7p13 y 15q15 para el TDAH o 1p36-p34, 2p16-p15, 11p15.5, 15q21.3 y Xq27.3 para la dislexia 1. Sin embargo, los estudios realizados mediante ligamiento, no son generalmente concordantes entre ellos, en la identificación de estos locus.
Los estudios de asociación en los TND son igualmente numerosos y en gran medida discordantes en sus resultados. La mayor parte de las variantes genéticas identificadas, si bien están significativamente asociadas a los TND, muestran odds ratios muy bajos, y algunas de ellas están presentes hasta en el 70% de la población, por lo que pueden considerarse moduladoras, no causales 1.
Empleando como ejemplo los GWAS (genome-wide association studies), estos precisan grandes poblaciones, test múltiples para evitar encontrar falsos positivos y tienden a mostrar asociaciones con variantes genéticas presentes en más de un 5% de la población, y por tanto, dudosamente causales 7. Estas variantes no son específicas de un TND, sino que pueden encontrase asociados a otros TND u otras enfermedades multisistémicas; la replicabilidad de estos resultados es adicionalmente muy baja.
Sin embargo, no debemos confundir los estudios de apoyo o rastreo genético encaminados al conocimiento o apoyo de la carga genética de los TND, con los estudios genéticos realizables en la práctica clínica para el diagnóstico causal del trastorno.
Estudios genéticos en la práctica clínica
Los estudios genéticos son un requerimiento habitual en numerosos trastornos del neurodesarrollo o enfermedades neurológicas. El especialista debe conocer las características y rentabilidad de las mismas según la patología a estudio. Ante la sospecha de un problema genético subyacente, la rentabilidad diagnóstica de los estudios genéticos, será generalmente paralela a la resolución de la misma.
Entre las técnicas citogenéticas debemos destacar el cariotipo, la hibridación in situ fluorescente (FISH) y la hibridación genómica comparada (CGH). El cariotipo permite la detección de anomalías estructurales superiores a 5Mb; el cariotipo de más de 450 bandas, obtenido a través de cromosomas prometafásicos, tipifica alteraciones cromosómicas con un tamaño superior a 3 Mb. La FISH y la CGH permiten el estudio de alteraciones estructurales de menor tamaño (2-10Mb), aunque con menor cobertura.
Las técnicas de genética molecular permiten el estudio de pequeñas alteraciones o mutaciones, y entre ellas debemos destacar los estudios de MLPA –multiplex ligation dependent probe amplification–, los de array (aCGH) y los de secuenciación. La primera, puede detectar anomalías de 50-70 pb; sin embargo, como en el FISH, se debe tener una alta sospecha de la patología y el origen de la misma dada su cobertura. La aCGH permite demostrar la presencia de deleciones o duplicaciones a lo largo de todo el genoma entre 1 y 5 Mb, aunque puede incrementarse su resolución para ver alteraciones menores de 25 kb; sin embargo, no detectan reordenamientos balanceados ni mosaicismos bajos. Dentro de los estudios por secuenciación debemos enfatizar la de un gen concreto, paneles de genes, del exoma clínico, y las exómica y genómicas masivas, individuales o en trío (analizando a padres); estas técnicas de alto rendimiento son capaces de realizar simultáneamente numerosas operaciones de secuenciación genética, y se las denomina genéricamente
como NGS o next-generation sequencing; éstas permiten análisis de 1bp, incrementando por 100 la cobertura diagnóstica respecto a los aCGH.
La indicación de los estudios genéticos es amplia.
La presencia de ciertas anomalías ecográficas o triple cribado alterado en la época prenatal, la existencia de defectos congénitos o genitales ambiguos en el neonato, la constatación de abortos de repetición, familiares de primer grado con trastornos genéticos y la presencia de talla patológica en el adolescente o adulto, pueden ser indicaciones para la realización de estudios genéticos.
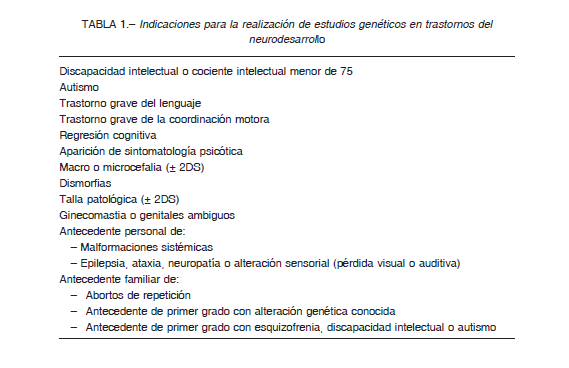
En los TND, la conveniencia de los estudios genéticos estará sujeta a la historia clínica (anamnesis adecuada y una exploración física completa) y al diagnóstico en sí. La existencia en el historial personal de malformaciones sistémicas o antecedentes familiares particulares (abortos de repetición, padre/hermano con alteración cromosómica, discapacidad intelectual o autismo), deben hacer plantear al clínico la conveniencia de los estudios genéticos. La exploración física puede poner de manifiesto signos compatibles con alteraciones genéticas que deberán valorarse detenidamente: dismorfias o defectos congénitos menores, talla patológica, alteraciones marcadas de crecimiento craneal, genitales ambiguos o ginecomastia. La indicación de estos estudios también está sujeta al tipo de trastorno. Así, la mayor parte de las guías recomienda la realización de estudios genéticos en pacientes con discapacidad intelectual (DI) o TEA 8. En la regresión cognitiva o la aparición de sintomatología psicótica, deberá contemplarse igualmente la necesidad de los estudios señalados, particularmente cuando el paciente había tenido previamente algún trastorno del neurodesarrollo. En otros TND se deberá contemplar la necesidad de estudios genéticos según la presencia de otras variables previamente señaladas (antecedentes o exploración física sugerente de genopatía) (Tabla 1).
Rentabilidad de los estudios genéticos
Podemos afirmar que la rentabilidad de los estudios genéticos en los TND, en el diagnóstico etiológico, es claramente superior a cualquier otra exploración complementaria.
Si tomamos como ejemplo la DI, la utilidad de los estudios metabólicos aporta la causa en el 0.2-7% (media 1%) y los estudios de neuroimagen muestran anomalías en el 30-40% de los casos, pero aportan el diagnóstico en el 0.2-3.9% de los casos 8. Estas cifras contrastan con la rentabilidad de las nuevas técnicas de secuenciación que demuestran la causa hasta en el 50% de los casos 8.
En la DI específicamente, la utilidad diagnóstica de los estudios genéticos va parejamente asociada a su resolución; así, las técnicas citogenéticas no superan el 6-10%, mientras que técnicas como la aCGH, la secuenciación exómica masiva (WES) o genómica masiva (WGS) en trío, alcanzan cifras del 15-23%, 24-33% y 55-70% respectivamente en la DI grave 9. Paradójicamente, la utilidad de estas técnicas genéticas en los TEA, parece mostrarse discretamente inferior ante la ausencia de DI asociada 10, 11; esto puede deberse a diferentes factores: diagnóstico más subjetivo e impreciso ante la ausencia de DI, presencia de factores ambientales o epigenéticos más condicionantes, mayor frecuencia de trastornos genéticos de doble impacto o de trastornos recesivos. En esta línea, podemos destacar el estudio mediante WES en trío en población saudí, donde la rentabilidad fue marcadamente elevada a expensas de trastornos autosómicos recesivo 12.
Los resultados del estudio meta-analítico sobre la utilidad de los estudios por aCGH, WES y WGS en niños con sospecha de enfermedades o trastornos genéticos es contundente. Con estas técnicas se demostró la causa del trastorno en el 10, 36 y 41% de los casos 13. La rentabilidad es dos veces superior cuando los estudios se realizan en trío, y alcanzan cifras hasta del 68% ante la presencia de rasgos dismórficos 14. La rentabilidad es igualmente superior ante el conocimiento técnico e implicación del clínico 13, 15. Cuando analizamos la utilidad de las pruebas por secuenciación, podemos objetivar una rentabilidad del 3-15% en estudios de gen único, 10-20% en paneles, más del 20% con la secuenciación exómica, más del 30% en la secuenciación exómica masiva en trío y más del 40% en la secuenciación genómica.
Dentro del apartado utilidad, no podemos obviar la evaluación del coste-efectividad. En pacientes con sospecha de trastornos genéticos, recurrir a las técnicas de secuenciación tras la realización de estudios básicos y complejos, frente a la realización inicial de estos estudios supone un incremento sustancial en el gasto (27 000 vs. 6000 AU$) 16; resultados similares se ha reportado en otros trabajos, que además apuntan la “odisea” de una demora en el diagnóstico causal de 6 años, 19 pruebas médicas previas y 8 consultas especializadas de media 17.
La rentabilidad diagnóstica, así como la económica, está condicionando los cambios en los algoritmos de diagnóstico genético. Hace 5 años, algunos protocolos recomendaban la NGS en pacientes con sospecha de procesos de origen genético, una vez confirmados los resultados negativos de la aCGH 18; un año después, se sugería sustituir el NGS por estudios de WES/WGS en trío; recientemente se ha propuesto la realización de NGS, antes incluso de la aCGH 19. A la vista de la rentabilidad de las pruebas genéticas moleculares, debemos igualmente plantear la conveniencia de estos estudios, previamente a la realización de estudios metabólicos o por neuroimagen 8.
Utilidad del diagnóstico etiológico
Debemos recalcar la relevancia del diagnóstico etiológico en el ejercicio médico, y por supuesto en los TND. La identificación de la causa del problema, podrá ayudar a anticipar problemas asociados, valorar si existe un tratamiento específico para el proceso base, evitar pruebas innecesarias, realizar los exámenes complementarios dirigidos según la causa, establecer estrategias preventivas, consejo genético, intervención terapéutica más útil según casuística previa, referir a grupos de apoyo especializados y establecer estrategias educativas planificadas a largo plazo entre otros aspectos (Tabla 2).
En esta misma línea, el estudio meta-analítico previamente mencionado arrojó una clara rentabilidad clínica en el 13, 17 y 27% de los estudios realizados mediante aCGH, WES y WGS respectivamente 13.
La resistencia a los estudios genético-moleculares está sujeta a diferentes factores. Los costes económicos, a pesar de los resultados mostrados. La presencia de resultados de significado incierto, circunstancia presente en cualquier exploración complementaria. La escasa ayuda al tratamiento o al diagnóstico, o la propia complejidad técnica, a pesar de los datos aportados en esta revisión.
De la utilidad a los modelos teóricos
Indudablemente, no podemos separar la genética del ambiente. Este último, desde su implicación en la genética hasta la participación en el propio desarrollo del sistema nervioso, va a modular la expresión de cualquier variante genética. Podemos observar cómo pacientes con la misma alteración genética, muestran patrones clínicos diferentes, sujetos a la participación del ambiente, e indudablemente al impacto de otros genes, ampliándose el concepto de la poligenia a la omnigenia.
Sin embargo, en estos modelos poligénicos, a la vista de los resultados mostrados, parece que los modelos poligénicos sumativos conviven con los modelos poligénicos multiplicativos 20. En el primero, la combinación de diferentes variantes genéticas con OR muy bajos, resultarían en la presencia de un TND; en el modelo multiplicativo (o de efecto mayor), una variante genética de OR elevado, sería la causa del TND, y otras variantes, así como el ambiente, serían moduladores del fenotipo final. A la vista de los resultados mostrados en la DI o TEA, este último modelo está cobrando un papel muy importante.
En conclusión, el diagnóstico etiológico en los TND va a permitir un mayor y mejor conocimiento de estos problemas. Dada la rentabilidad de los estudios genéticos, particularmente en la DI y el TEA, debemos acometer estos estudios tempranamente. El diagnóstico causal podrá facilitar el asesoramiento personalizado en el patrón de recurrencia, y la posible anticipación de enfermedades multisistémicas entre otros aspectos; podrá igualmente contribuir en el entendimiento del problema del paciente, evitar pruebas innecesarias, y eliminar teorías de pobre sustento científico, aspectos todos trascendentales para el paciente y su familia.

La complejidad de estas nuevas técnicas condiciona la formación de los especialistas que atienden estos problemas, para la obtención de una mayor rentabilidad.
Conflicto de intereses: Ninguno para declarar
Bibliografía
1. Fernández-Jaén A, Cigudosa JC, Martín Fernández-Mayoralas D, et al. Genética aplicada a la práctica clínica en trastornos del neurodesarrollo. Rev Neurol 2014; 58 (Suppl 1): S65-70.
2. Fernandez-Jaen A, Fernandez-Mayoralas DM, Calleja-Perez B, Munoz-Jareno N, Lopez-Arribas S. Endofenotipos genomicos del trastorno por deficit de atencion/hiperactividad. Rev Neurol 2012; 54 (Suppl 1): S81-7.
3. Ropers HH. Genetics of early onset cognitive impairment. Annu Rev Genomics Hum Genet 2010; 11: 161-87.
4. Hallmayer J, Cleveland S, Torres A, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry 2011; 68: 1095-102.
5. Astrom RL, Wadsworth SJ, DeFries JC. Etiology of the stability of reading difficulties: the longitudinal twin study of reading disabilities. Twin Res Hum Genet 2007; 10: 434-9.
6. Larsson H, Chang Z, D’Onofrio BM, Lichtenstein P. The heritability of clinically diagnosed attention deficit hyperactivity disorder across the lifespan. Psychol Med 2014; 44: 2223-9.
7. Koeleman B, Al-Ali A, van der Laan S, Asselbergs F. A concise history of genome-wide association studies. Saudi J Med Med Sci 2013; 1: 4-10.
8. Fernández-Jaén A, López-Martín S, Albert J, Martín D, Calleja-Pérez B. Evaluación, diagnóstico etiológico y planteamiento terapéutico. En: Fernández-Jaén A, Fernández-Perrone A, Martín D, editors. Trastornos del neurodesarrollo. Discapacidad intelectual y trastornos de la comunicación. Madrid, España: Editorial Médica Panamericana, 2019, p 61-104.
9. Vissers LELM, Gilissen C, Veltman JA. Genetic studies in intellectual disability and related disorders. Nature Reviews Genetics 2016; 17: 9-18.
10. Ji J, Shen L, Bootwalla M, et al. A semiautomated wholeexome sequencing workflow leads to increased diagnostic yield and identification of novel candidate variants. Cold Spring Harb Mol Case Stud 2019; 5: doi: 10.1101/mcs.a003756.
11. Robert C, Pasquier L, Cohen D, et al. Role of Genetics in the Etiology of Autistic Spectrum Disorder: Towards a Hierarchical Diagnostic Strategy. Int J Mol Sci 2017; 18: doi: 10.3390/ijms18030618.
12. Al-Mubarak B, Abouelhoda M, Omar A, et al. Whole exome sequencing reveals inherited and de novo variants in autism spectrum disorder: a trio study from Saudi families. Sci Rep 2017; 7: 5679.
13. Clark MM, Stark Z, Farnaes L, et al. Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. NPJ Genom Med 2018; 3: 16.
14. Bick D, Jones M, Taylor SL, Taft RJ, Belmont J. Case for genome sequencing in infants and children with rare, undiagnosed or genetic diseases. J Med Genet 2019; 56: 783-91.
15. Mak CC, Leung GK, Mok GT, et al. Exome sequencing for paediatric-onset diseases: impact of the extensive involvement of medical geneticists in the diagnostic odyssey. NPJ Genom Med 2018; 3: 19.
16. Stark Z, Schofield D, Alam K, et al. Prospective comparison of the cost-effectiveness of clinical whole-exome sequencing with that of usual care overwhelmingly supports early use and reimbursement. J Genet Med 2017; 19: 867.
17. Tan TY, Dillon OJ, Stark Z, et al. Diagnostic impact and cost-effectiveness of whole-exome sequencing for ambulant children with suspected monogenic conditions. JAMA Pediatr 2017; 171: 855-62.
18. Shashi V, McConkie-Rosell A, Rosell B, et al. The utility of the traditional medical genetics diagnostic evaluation in the context of next-generation sequencing for undiagnosed genetic disorders. J Genet Med 2014; 16: 176-82.
19. Srivastava S, Love-Nichols JA, Dies KA, et al. Meta-analysis and multidisciplinary consensus statement: exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. J Genet Med 2019; 21: 2413-21.
20. Berg JM, Geschwind DH. Autism genetics: searching for specificity and convergence. Genome Biol 2012; 13: 247.