
BEATRIZ OLIVERI 1, DIANA C. GONZÁLEZ 2, PAULA ROZENFELD 3, EMMA FERRARI 4, GLADYS GUTIÉRREZ 5
1 Laboratorio de Osteoporosis y Enfermedades Metabólicas Óseas, Instituto de Inmunología, Genética y Metabolismo (INIGEM), Facultad de Farmacia y Bioquímica, Hospital de Clínicas José de San Martin, UBA-CONICET, Buenos Aires, 2 Mautalén, Salud e Investigación, Buenos Aires, 3 IIFP, Departamento de Ciencias Biológicas, Facultad de Ciencias Exactas-CONICET, Universidad Nacional de La Plata, La Plata, 4 Hospital Samco, Villa Ocampo, Santa Fe, 5 Servicio de Hemato-oncología, Hospital Juan Pablo II, Corrientes, Argentina
En representación del Grupo de estudio Bone Involvement Gaucher Disease (BIG)
Resumen La enfermedad de Gaucher (EG) es causada p or una deficiencia genética de la glucocerebrosidasa (GCasa) que provoca acumulación de glucocerebrósido en hígado, bazo y médula ósea. La terapia temprana de reemplazo enzimático revierte citopenias, visceromegalias y previene lesiones óseas irreversibles, por lo cual el diagnóstico precoz es fundamental. Los algoritmos diagnósticos en uso apuntan a manifestaciones hematológicas clásicas. Los síntomas óseos están presentes en 25-32% de los pacientes pero no suelen despertar sospecha de EG. Diseñamos un programa educativo sobre la afecta ción ósea de la EG y un algoritmo focalizado en la presentación con manifestaciones óseas para facilitar su diagnóstico precoz (proyecto BIG: Bone Involvement in Gaucher Disease). El objetivo del trabajo es describir el proyecto BIG y los resultados de su aplicación en nuestra consulta. Entre marzo de 2017 y diciembre de 2018 se recibieron 38 muestras de sangre seca de pacientes con alguna manifestación ósea sospechosa de EG para cuantificar la actividad de GCasa. Una muestra no cumplía los criterios de inclusión y en 3 de las 37 restantes se observó actividad deficiente de GCasa. El diagnóstico de EG se confirmó por medición de GCasa en leucocitos en dos niñas con manifestaciones óseas de 4 y 2 años de evolución, respectivamente, sin citopenia ni visceromegalia clínicamente evidentes. En el otro paciente con baja actividad la medición en leucocitos fue normal. Los casos detectados muestran la efectividad de un programa educacional de difusión y la utilidad de un algoritmo de detección precoz basado en síntomas óseos que facilitaría el diagnóstico de EG.
Palabras clave: enfermedad de Gaucher, diagnóstico precoz, algoritmo diagnóstico, glucocerebrosidasa, determinación en sangre seca
Abstract Gaucher disease (GD) is caused by a genetic deficiency of the lysosomal enzyme glucocerebrosidase (GCase) leading to the accumulation of glucocerebroside in the liver, spleen, and bone marrow. The early diagnosis allows a prompt enzyme replacement therapy reversing cytopenias and visceromegaly and preventing irreversible bone lesions. Current diagnostic algorithms are based on well-recognized hematological manifestations. Although bone symptoms are present in 25-32% of the patients, they are not usually suspected as associated with Gaucher disease at clinical presentation. We designed an educational program aimed to give advice on the skeletal involvement in GD and a new diagnostic algorithm that considers bone symptoms to facilitate its early diagnosis (BIG project: Bone Involvement in Gaucher Disease). The study aims at describing the BIG project and the results of its application in our clinic in various cities in Argentina. Within the frame of this project, between March 2017 and December 2018, 38 dry blood spot samples from patients with bon e manifestations suspected of having GD were submitted to quantification of GCase activity. One sample did not meet the inclusion criteria. Deficient GCase activity was detected in three of the remaining 37 samples. The diagnosis of GD was confirmed in two girls who presented bone manifestations of 4 and 2 years of evolution, respectively, without hematological alterations. The third patient with low enzyme activity had normal leukocyte GCase. The two newly diagnosed cases of GD show the efficacy of our dual strategy aimed to facilitate the early diagnosis of this rare disease.
Key words: Gaucher disease, early diagnosis, algorithms, glucocerebrosidase, dried blood spot testing
Dirección postal: Beatriz Oliveri, Laboratorio de Osteoporosis y Enfermedades Metabólicas Óseas, Instituto de Inmunología, Genética y Metabolismo (INIGEM), Facultad de Farmacia y Bioquímica, Hospital de Clínicas José de San Martín, UBA-CONICET, Córdoba 2351, 1120 Buenos Aires, Argentina
e-mail: beatrizoliveri258@gmail.com
Conocimiento actual
• Los algoritmos diagnósticos actualmente en uso están basados en las manifestaciones hematológicas clásicas.
Las manifestaciones óseas son menos reconocidas lo que puede causar demoras en el diagnóstico.
• El tratamiento temprano de la enfermedad de Gaucher revierte las citopenias y visceromegalias y previene lesiones óseas irreversibles.
Contribución al conocimiento actual
• Presentamos un programa educativo y un algoritmo focalizado en síntomas óseos que facilitaron el reconocimiento de la enfermedad en dos pacientes con sintomatología ósea sin manifestaciones hematológicas ni visceromegalias significativas.
La enfermedad de Gaucher (EG) es una de las enfermedades lisosomales más frecuentes. Es causada por mutaciones del gen GBA1 que codifica la síntesis de la enzima lisosomal glucocerebrosidasa (GCasa). La ausencia o deficiencia de GCasa produce la acumulación de glucosilceramida en los lisosomas del sistema monocítico macrofágico de médula ósea, bazo, hígado, con menor frecuencia en pulmones y, en las formas neurológicas, en cerebro, causando daño celular y disfunción orgánica 1.
El fenotipo más frecuente es la EG tipo I (EG1), con manifestaciones principalmente hematológicas, viscerales y esqueléticas. Su frecuencia varía desde 1:40 000 a 1:60 000 en la población general, y alcanza 1:800 en la población judía Ashkenazi 1.
La sospecha clínica de EG1 está tradicionalmente asociada a la presencia de espleno/hepatomegalia y citopenias, por lo cual los algoritmos de diagnóstico están elaborados sobre la base de dichos signos y son los utilizados generalmente por los hematólogos 2-4. Sin embargo, se ha observado que hasta 25-32% de los pacientes presentan signos y/o síntomas óseos como manifestación inicial de la enfermedad 5-8.
El 80%-95% de los pacientes con EG1 (incluso los asintomáticos) tienen afectación ósea al momento del diagnóstico que puede incluir alguna o varias de las siguientes características: deformación en frasco de Erlenmeyer, disminución de densidad mineral ósea, infartos óseos, necrosis ósea avascular, lesiones radiológicas osteolíticas o esclerosantes y fracturas. El 27- 63% refiere antecedentes de dolor óseo y el 9-20% antecedentes de crisis óseas lo que demostraría que el retraso en el diagnóstico permite el avance de la afectación ósea 3, 5-9. Tratándose de una enfermedad poco frecuente, el desconocimiento o la falta de alerta sobre las posibles manifestaciones óseas iniciales puede provocar demora en el diagnóstico y por lo tanto en el inicio del tratamiento específico, dando lugar a severas complicaciones óseas que resultan irreversibles y que alteran la funcionalidad, movilidad y la calidad de vida de estos pacientes 5, 8, 10.
Desde 1991, existen terapias de reemplazo enzimático basadas en la infusión de GCasa. Los fármacos disponibles y aprobados en la Argentina, velaglucerasa alfa e imiglucerasa recombinante, mejoran sustancialmente las citopenias, visceromegalia, el crecimiento y el pico de masa ósea en los niños. El tratamiento de reemplazo enzimático administrado precozmente, con dosis y adherencia adecuadas, puede prevenir las complicaciones óseas irreversibles 11-14.
Considerando la importancia del diagnóstico temprano, desarrollamos el Proyecto BIG (Bone Involvement in Gaucher Disease) que incluye un programa educativo, un algoritmo de detección precoz basado en manifestaciones óseas sospechosas de EG 15 y charlas de difusión en diferentes ámbitos científicos. El objetivo de este trabajo es describir el proyecto BIG y los resultados de su aplicación desde 2017 en Argentina. Para ejemplificar las dificultades referidas en el diagnóstico, se incluye además una breve reseña de dos pacientes con EG diagnosticados como resultado de la activa difusión para el reconocimiento de la enfermedad como se plantea en el programa diseñado a tal fin.
Materiales y métodos
Las muestras de pacientes fueron derivados al laboratorio siguiendo dos rutas diferentes, a saber:
1. Protocolo 1 (BIG algoritmo): El programa BIG consta de sesiones educativas que se desarrollaron en hospitales y centros médicos del país con asistencia de profesionales de especialidades relacionadas con las posibles manifestaciones clínicas de la EG incluyendo la patología ósea. Cada sesión consta de un módulo intensivo de fisiopatología, diagnóstico, afectación ósea y tratamiento de EG, con entrenamiento en el uso del algoritmo diseñado para diagnóstico precoz de EG basado en manifestaciones óseas y en sus procedimientos (ficha clínica, consentimiento informado, obtención y envío de muestras) (Fig. 1). Los signos y síntomas que deben alertar al profesional se describen en la Tabla 1. Ante la presencia de alguno de los signos/síntomas de los grupos A y B de la Tabla 1, se completa una ficha clínica, y previa obtención consentimiento informado, se envía una muestra de gotas de sangre seca (DBS, por dried blood sample), para medición de la actividad de GCasa 16, 17. A los fines de un estudio de tamizaje, esta determinación es ventajosa porque requiere unas pocas gotas de sangre en papel de filtro que puede enviarse por correo a temperatura ambiente. Sin embargo, la actividad enzimática deficiente en DBS no es confirmatoria de EG por lo cual el diagnóstico requiere la determinación de la actividad de la enzima GCasa en leucocitos aislados de sangre periférica y/o fibroblastos en cultivo. La medición de GCasa en leucocitos se realiza en una muestra de sangre colectada con heparina, que debe enviarse refrigerada y en un tiempo no mayor a 24 horas. Por lo tanto, si el resultado en DBS muestra actividad enzimática reducida, se solicita la medición de la enzima en leucocitos circulantes y un hemograma con recuento de plaquetas y dosaje de ferritina.
En aquellos pacientes con signos del Grupo C, se solicita primero el hemograma con recuento de plaquetas y dosaje de ferritina. Si alguno de estos resultados es anormal, se solicitará la determinación de actividad enzimática en DBS, y si el resultado es positivo para EG se confirma el diagnóstico con la medición de la enzima en leucocitos circulantes. La actividad normal de GCasa ya sea por DBS o medida en leucocitos circulantes descarta EG. En cada centro se nombró un coordinador responsable de verificar que se cumplieran los procedimientos para la obtención y envío de muestras para medición de actividad GCasa en gota seca y en leucocitos y que se entreguen los resultados al médico derivador para que por su intermedio lleguen a los pacientes adultos o los padres y/o tutores de pacientes menores de edad.
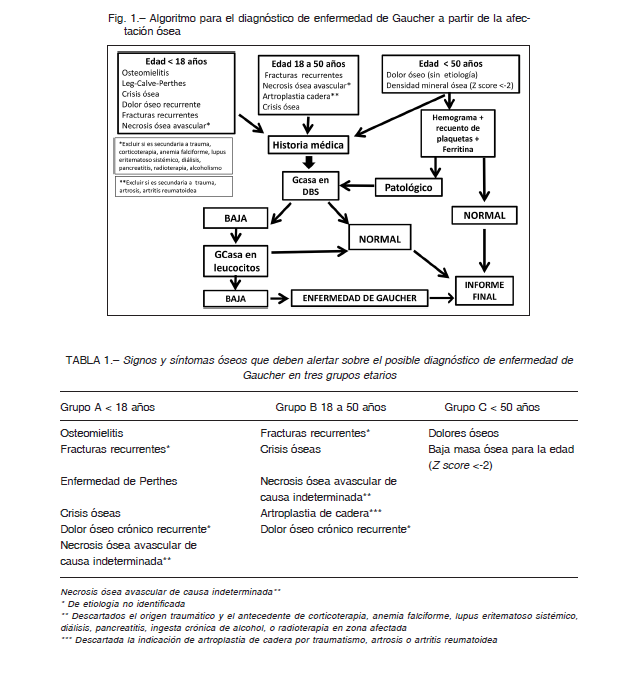
2. Protocolo 2 (BIG docente): sesiones educativas con enfoque similar al protocolo BIG base dictadas en congresos, jornadas, cursos, ateneos de sociedades médicas y/o servicios relacionados con enfermedades óseas invitando a los médicos asistentes a derivar muestras de DBS de aquellos pacientes con sospecha clínica de EG.
Se obtuvo consentimiento informado escrito de las dos pacientes cuyos datos se presentan y de t odos los pacientes a los que se solicitó muestra para medición de actividad GCasa.
Resultados
Entre marzo de 2017 y diciembre de 2018 se realizaron 12 reuniones educativas en hospitales y centros de salud de Argentina: 4 en la Ciudad de Buenos Aires y alrededores, el resto en el interior del país (provincias de Córdoba, Corrientes, Chaco, Jujuy, La Pampa, Mendoza, San Juan y Santa Fe, ver Apéndice 1). Asistieron 321 profesionales, en su mayoría pediatras, hematólogos, traumatólogos y reumatólogos (79% de los asistentes), y también endocrinólogos, radiólogos, especialistas en errores innatos del metabolismo, clínicos, osteólogos, bioquímicos y nefrólogos.
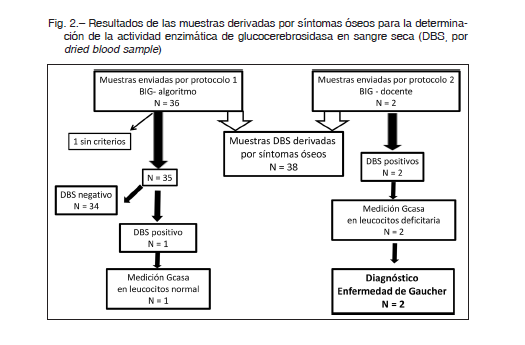
Por el protocolo 1, se recibieron muestras de DBS de 36 pacientes. Un paciente no cumplía los criterios de inclusión. Los pacientes restantes (23 mujeres, 12 varones) tenían edad promedio en años: 15.3 (rango 1.4-69.0); mediana 8.5; IQR 12.25. El 77% era menor de 20 años. Las principales causas de derivación fueron dolor o crisis óseas (77%), fracturas (26%), necrosis ósea avascular (17%) y presunta osteomielitis (3%); los porcentajes parciales no suman 100% porque algunos pacientes tenían más de un síntoma. El 46% presentaba síntomas óseos exclusivamente, mientras que en el 54% restante se acompañaban de alguna de las manifestaciones: citopenias, visceromegalias, retardo de crecimiento, HDL bajo, litiasis vesicular o fatiga crónica. De las 35 muestras de DBS, 34 presentaron actividad normal de GCasa. En la muestra restante, la actividad GCasa fue deficiente, sin embargo el estudio de GCasa en leucocitos resultó normal.
Por el protocolo 2, se recibieron muestras de DBS de 2 pacientes. En ambas muestras fue confirmada la deficiencia por medición en leucocitos circulantes, arribando al diagnóstico de EG y ambas pacientes comenzaron tratamiento de reemplazo enzimático con velaglucerasa (Fig. 2). Los datos clínicos y otras características de interés de las dos pacientes se muestran en la Tabla 2.
Caso 1: Niña que comenzó a los 7 años con dolor en rodilla y diagnóstico de fractura de stress femoral. A los 10a y 4m y a los 11a y 2m presentó nuevos episodios de dolor intenso en rodilla derecha y tercio distal de fémur izquierdo acompañados de signos de flogosis, eritrosedimentación y PCR elevados, leve anemia y leucocitosis.
Fueron interpretados y tratados como osteomielitis a pesar de los cultivos negativos. Se descartó enfermedad oncológica por biopsia. A raíz de las sospechas del grupo multidisciplinario tratante y la sugerencia de una médica reumatóloga que había asistido a una de las charlas de difusión, se envió una muestra para dosaje de GCasa en DBS que mostró deficiencia de la enzima, confirmada por el dosaje en leucocitos. Los niveles de biomarcadores de EG estaban aumentados (quitotriosidasa, ferritina, ECA) y no presentaba citopenias o visceromegalias significativas.
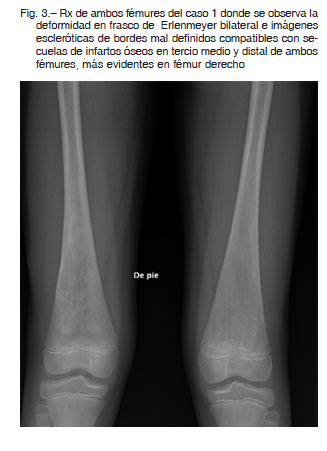
En ambos fémures se observaba deformación en frasco de Erlenmeyer (inadvertido en las Rx a los 7 años de edad) y lesiones compatibles con infartos óseos (Fig. 3). La resonancia magnética nuclear de ambos fémures mostró infiltración de médula ósea e infartos óseos en las áreas coincidentes con la sintomatología interpretada como osteomielitis a los 10 y 11 años. No presentaba alteraciones del laboratorio óseo y mineral y su densitometrías óseas eran normales (densidad mineral ósea columna lumbar L1-L4: Z- score – 0.9 y esqueleto total: Z-score -1.1).
Caso 2: niña de 9 años con dolor en miembro inferior derecho, de dos años de evolución sin diagnóstico. Sin citopenias ni visceromegalias clínicas y, como en el caso anterior, consultó a diferentes especialistas, hasta que la hemato-oncóloga, luego de descartar otras patologías, sospechó EG por haber recibido información sobre las manifestaciones óseas en esta enfermedad. Se envió una muestra para dosaje de GCasa en DBS que mostró deficiencia de la enzima, luego confirmada por el dosaje en leucocitos. Al momento del diagnóstico de EG, la paciente no presentaba alteraciones del laboratorio óseo y mineral y su densitometría ósea era normal (L1-L4: Z-score -0.2 y esqueleto total: Z-score -0.3). En la resonancia magnética nuclear de ambos fémures se observó infiltración ósea en las áreas de dolor.

Discusión
La presencia de síntomas óseos como manifestación inicial de la EG tipo I es mucho menos sospechada que las alteraciones hematológicas y visceromegalias. Por este motivo no es infrecuente el retraso en el diagnóstico de EG en esos pacientes sin alteraciones hematológicas clínicamente significativas. Rossi y col. evaluaron un grupo de 44 pacientes con manifestación ósea inicial (41 con EG tipo I) que presentaron los primeros síntomas antes de los 16 años y encontraron que entre el primer síntoma y el diagnóstico había transcurrido un tiempo promedio de 4 años (rango 0-36 años) y que el 32% de los pacientes refirió haber tenido de inicio alguna de las siguientes manifestaciones: necrosis ósea aséptica, infartos óseos, dolor o crisis óseas, (uno había sido erróneamente diagnosticado como osteomielitis de la cadera) e incluso en el 18% la manifestación única inicial fue ósea6. En un grupo de adultos con EG, el 25% refirió haber presentado inicialmente síntomas óseos, crisis o dolor óseo 7.
Los dos casos que, a modo de ejemplo, se describen en la Tabla 2, coinciden con la bibliografía con respecto a la dificultad en el diagnóstico, como lo demuestran el tiempo de demora, el número de profesionales consultados, la variedad de especialidades involucradas y los diagnósticos erróneos como el de osteomielitis.
Es interesante remarcar el fémur en matraz de Erlenmeyer que pasó inadvertido en el caso 1 en una radiografía realizada 4 años antes del diagnóstico. Esta imagen característica es un hallazgo frecuente en la EG, y corresponde a la expansión de la médula ósea por infiltración de células de Gaucher con la consecuente alteración del modelamiento óseo 10. Es posible pensar entonces que el primer episodio de fractura sufrido por la niña a los 7 años fue consecuencia de un hueso debilitado por la infiltración de células de Gaucher, y que los posteriores episodios de dolor intenso presentados por la paciente en ambos fémures se trataron de crisis óseas. Estas se caracterizan por ser cuadros agudos de dolor inicialmente sordo que progresa hasta volverse intenso en 2 a 3 días y puede persistir por 7 a 10 días 10. Generalmente se acompañan de signos de inflamación local, leucocitosis y eritrosedimentación elevada, con o sin fiebre, por lo cual pueden confundirse fácilmente con osteomielitis 18, 19. Durante la crisis ósea de la EG, sin embargo, no hay toxemia y los cultivos son negativos, como ocurrió en esta paciente. El centellograma óseo muestra hipercaptación en la osteomielitis a diferencia de lo que ocurre en la crisis óseas de EG donde el hueso no hipercapta el trazador 18.
En el caso 2, la sintomatología principal fue dolor óseo recurrente en miembro inferior derecho, probablemente asociado a la infiltración de médula ósea en ambos fémures, a predominio en derecho como se observa en la resonancia magnética nuclear, lo cual coincide con el patrón de comienzo de la infiltración en las diáfisis de huesos largos, siendo fémures y tibias los más afectados.
Cabe destacar que una de las metas del tratamiento de reemplazo enzimático es disminuir o eliminar el dolor óseo 20.
El cuadro clínico de dolor en las dos pacientes fue inicialmente confuso ya que no se acompañó de citopenias o visceromegalias significativas, que son los otros componentes característicos y clásicamente más reconocidos de esta enfermedad. En el análisis de ambos casos se identifican signos óseos que podrían haber alertado tempranamente sobre la presencia de una EG y que son los propuestos en el algoritmo diagnóstico del proyecto 1 BIG 15. La paciente 1 presentaba dos criterios de inclusión en el algoritmo: “osteomielitis” sin identificación de causa y crisis óseas y la paciente 2 dolor óseo de prolongada evolución, recurrente y sin causa aparente. La advertencia de dichos criterios permite la sospecha diagnóstica y el envío de las muestras para evaluar actividad de GCasa.
Los algoritmos hasta ahora propuestos para diagnóstico precoz de EG están basados principalmente en sintomatología hematológica 2-4. Motta y colaboradores mostraron resultados de su aplicación por un periodo de 39 meses en 34 centros de hematología de Italia 3. A partir de pacientes con esplenomegalia y/o trombocitopenia, luego de descartar las causas comunes, se enviaron 196 muestras de DBS (edad promedio 47.8 años). La actividad GCasa en DBS resultó deficiente en 34 de ellas. A estos pacientes más uno con DBS negativo pero con historia familiar de EG, se les midió GCasa en homogenato celular (leucocitos, linfoblastos o fibroblastos). De 35 muestras recibidas, en 7 se confirmó diagnóstico de EG (edad promedio: 31.8 años). Por tanto, la prevalencia de diagnóstico positivo en dicha población seleccionada fue 3.6%.
En nuestro estudio, en un período de 21 meses, recibimos por ambos protocolos, 37 muestras con criterio de inclusión para DBS (edad promedio 15.5 años) de las cuales 3 mostraron actividad deficiente de GCasa y en dos se confirmó diagnóstico de EG por estudio en leucocitos, lo que representa una prevalencia en conjunto del 5.7% (con edad al diagnóstico 10.7 años). La diferencia en la edad entre los pacientes referidos en nuestro proyecto y el proyecto italiano estuvo seguramente relacionada con el mayor número de hospitales pediátricos en el grupo BIG.
En conclusión, consideramos que ambos casos ejemplifican el valor de la difusión del conocimiento de la EG y la probable utilidad del algoritmo a partir de manifestaciones óseas, incluido en el Proyecto BIG, que facilitaría la detección precoz de la enfermedad de Gaucher en los pacientes con predominio de síntomas óseos. Cabe puntualizar que el acceso a un tratamiento temprano evitaría las complicaciones esqueléticas irreversibles e incapacitantes que frecuentemente presentan estos pacientes en la evolución natural de la enfermedad.
Agradecimientos: El presente proyecto fue realizado en el marco de un subsidio para Educación Médica de Shire GmBh.
Conflicto de intereses: Las Dras. Beatriz Oliveri y Diana González han recibido de Shire honorarios docentes y becas para participar de congresos.
Paula Rozenfeld ha recibido honorarios docentes y apoyo para viajes y subsidios de investigación de Shire. El resto de las autoras, ninguno para declarar.
Bibliografía
1. Stirnemann J, Belmatoug N, Camou F, et al. A review of Gaucher disease pathophysiology, clinical presentation and treatments. Int J Mol Sci 2017; 18: pii: E441.
2. Mistry PK, Cappellini MD, Lukina E, et al. A reappraisal of Gaucher disease -diagnosis and disease management algorithms. Am J Hematol 2011; 86: 110-5.
3. Motta I, Filocamo M, Poggliali E, et al. A multicentre observational study for early diagnosis of Gaucher disease in patients with Splenomegaly and/or thrombocytopenia. Eur J Haematol 2016; 96: 352-9.
4. Di Rocco M, Andria G, Deodato F, Giona F, Micalizzi C, Pession A. Early diagnosis of Gaucher disease in pediatric patients: proposal for algorithm. Pediatr Blood Cancer 2014; 61: 1905-9.
5. Mistry PK, Sadan S, Yang R, Yee J, Yang M. Consequences of diagnostic delays in type 1 Gaucher disease: the need for greater awareness among hematologistsoncologists and an opportunity for early diagnosis and intervention. Am J Hematol 2007; 82: 697-701.
6. Rossi L, Zulian F, Stirnemann J, de Villemur TB, Belmatoug N. Bone involvement as presenting sign of pediatric-onset Gaucher disease. Joint Bone Spine 2011; 78: 70-4.
7. Oliveri B, González D, Quiroga, F, Silva C, Rozenfeld P. A comprehensive study of bone manifestations in adult Gaucher Disease type 1 patients in Argentina. Calcif Tissue Int 2019; 104: 1-8.
8. Thomas AS, Mehta AB, Hughes DA. Diagnosing Gaucher disease: an on-going need for increased awareness amongst haematologists. Blood Cells Mol Dis 2013; 50: 212-7.
9. Charrow J, Andersson H, Kaplan P et al. The Gaucher registry. Demographics and disease characteristics of 1668 patients with Gaucher disease. Arch Intern Med 2000; 160: 2835-43.
10. Mikosch P, Hughes D. An overview on bone manifestations in Gaucher disease. Wien Med Wochenschr 2010; 160: 609-24.
11. Elstein D, Foldes AJ, Zahrieh D, et al. Significant and continuous improvement in bone mineral density among type 1 Gaucher disease patients treated with velaglucerase alfa: 69-month experience, including dose reduction. Blood Cells Mol Dis 2011; 47: 56-61.
12. van Dussen L, Biegstraaten M, Dijkgraaf MG, Hollak CE. Modelling Gaucher disease progression: long-term enzyme replacement therapy reduces the incidence of splenectomy and bone complications. Orphanet J Rare D 2014; 9: 112.
13. Andersson H, Kaplan P, Kacena K, Yee J. Eight-year clinical outcomes of long-term enzyme replacement therapy for 884 children with Gaucher disease type 1. Pediatrics 2008; 122, 1182-90.
14. Khan A, Hangartner T, Weinreb NJ, Taylor JS, Mistry PK. Risk factors for fractures and avascular osteonecrosis in type 1 Gaucher disease: a study from the International Collaborative Gaucher Group (ICGG) Gaucher Registry. J Bone Min Res 2012; 27: 1839-48.
15. Rozenfeld P, Gonzalez D, Lis C, Riemersa O, Kot M, Oliveri B. BIG Project (Bone Involvement in Gaucher Disease): Screening for Gaucher disease with focus in bone affection. J Inborn Errors Metab Screen 2017; 5: 305.
16. Chamoles NA, Blanco MB, Gaggioli D, Casentini C . Gaucher and Niemann-Pick diseases – Enzymatic diagnosis in dried blood spots on filter paper: retrospective diagnoses in newborn screening cards. Clin Chim Acta 2002; 317: 191-7.
17. Ceci R, de Francesco PN, Mucci JM, Cancelarich NL, Fossati CA, Rozenfeld PA. Reliability of enzyme assays in dried blood spots for diagnosis of 4 lysosomal storage disorders. Adv Biochem 2011; 1: 58-64.
18. Baris HN, Weisz Hubshman M, Bar-Sever Z, Kornreich L, Shkalim Zemer V, Cohen IJ. Re-evaluation of bone pain in patients with type 1 Gaucher disease suggests that bone crises occur in small bones as well as long bones. Blood Cells Mol Dis 2016; 60: 65-72.
19. Paonessa KJ, McInerney VK, Minnefor AB. Pseudo-osteomyelitis in Gaucher’s disease. Orthop Rev 1989; 18: 880-8.
20. Drelichman G, Fernández Escobar N, Basack N, et al. Actualización del Consenso Argentino de Enfermedad de Gaucher: Grupo Argentino para el diagnóstico y tratamiento de la Enfermedad de Gaucher. Hematología 2015; 19: 4-51.
Apéndice 1
Grupo de estudio Bone Involvement Gaucher Disease (BIG)
Hospitales y Centros de Salud donde se desarrollaron las reuniones educativas BIG y sus coordinadores
Hospital Nacional Alejandro Posadas, Provincia de Buenos Aires: Dra. C. Riccheri
Hospital de Niños Víctor J. Vilela, Rosario, Santa Fe: Dra. A. Schifino
Hospital de Niños Santísima Trinidad, Cdad. Córdoba, Córdoba: Dr. N. Guelbert
Hospital Pediátrico Dr. Avelino Lorenzo Castelan, Resistencia, Chaco: Dra. C. Picon
Hospital de Niños Dr. Héctor Quintana, San Salvador de Jujuy, Jujuy: Dra. M. Tanus
Hospital de Pediatría SAMIC Prof. Dr. Juan P. Pedro Garrahan, CABA: Dras. A. Feliu y V. Avalos
Hospital de Niños Pedro de Elizalde, CABA: Dra. G. O. Elena
Hospital General Lucio Molas, Santa Rosa, La Pampa: Dras. N. Martin y B. Espinosa
Hospital Sczestakow, San Rafael, Mendoza: Dra. O. Ciprian
Mautalen Salud e Investigación, CABA: Dras. D. González y B. Oliveri
Hospital Notti, Cdad. Mendoza, Mendoza: Dr. G. Arbesu
Hospital Rawson, Cdad. San Juan, San Juan: Dra. V. Canónico