
MARÍA V. PAOLINI1, SILVIA DANIELIAN2, EMMA PRIETO2, MARÍA FERNANDA TAMI1,
MATÍAS M. OLEASTRO3, DIEGO S. FERNÁNDEZ ROMERO1
1Unidad Inmunología e Histocompatibilidad, Hospital Dr. Carlos G. Durand, 2Área de Biología Molecular,
Servicio de Inmunología y Reumatología, Hospital de Pediatría Prof. Dr. Juan P. Garrahan,
3Servicio de Inmunología, Hospital Prof. Dr. Juan P. Garrahan, Buenos Aires, Argentina
Resumen El síndrome WHIM es una inmunodeficiencia primaria de herencia autosómica dominante, debida
a mutaciones en el gen CXCR4, que se caracteriza por verrugas cutáneo-mucosas, hipogammaglobulinemia, infecciones bacterianas recurrentes y mielocatesis. El tratamiento se basa en el uso de antibióticos profilácticos, gammaglobulina en dosis sustitutiva y factores estimulantes de colonias de granulocitos o de granulocitos y macrófagos, en forma crónica. Presentamos el caso de una mujer de 21 años que comenzó a los 10 meses de edad con leucopenia y al siguiente año múltiples infecciones con hipogammaglobulinemia requiriendo gammaglobulina endovenosa durante los episodios. Evolucionó con neutropenia crónica. Una punción aspiración de médula ósea mostró la serie mieloide aumentada con ligero predominio de elementos inmaduros. El cuadro fue interpretado como inmunodeficiencia común variable debido a la asociación de múltiples cuadros infecciosos, niveles disminuidos de IgG, IgM e IgA y linfopenia con disminución de linfocitos B de memoria, por lo que comenzó tratamiento sustitutivo con gammaglobulina endovenosa más antibióticos profilácticos. A los 20 años se registraron pequeñas verrugas en manos que progresaron hacia antebrazos, abdomen, cara y rodillas. Se realizaron estudios moleculares para la búsqueda de mutaciones en el gen CXCR4 donde se detectó la mutación p.Arg334STOP en estado heterocigota confirmando el diagnóstico de síndrome WHIM, que es una inmunodeficiencia infrecuente y de difícil diagnóstico.
Palabras clave: inmunodeficiencia, WHIM, CXCR4, mielocatesis
Abstract Late diagnosis of WHIM syndrome. WHIM syndrome is a primary autosomal dominant immuno
deficiency due to CXCR4 mutations characterized by mucocutaneous warts, hypogammaglobulinemia, recurrent bacterial infections and myelokathesis. Treatment consists in prophylactic antibiotics, immunoglobulin replacement and granulocyte or granulocyte/monocyte colony stimulating factors. We present the case of a 21 year old woman who showed leukopenia at 10 months of age and one year later multiple infections with hypogammaglobulinemia requiring intravenous immunoglobulin. During follow up she developed chronic neutropenia. A bone marrow aspiration showed increased myeloid series with predominance of immature elements. On the basis of infections, low levels of IgG, IgA, IgM and lymphopenia with absent memory B cells, a diagnosis of common variable immunodeficiency was made. She started intravenous immunoglobulin replacement and prophylactic antibiotics. At age 20, small warts in hands that progressed to forearms, knees, abdomen and face were recorded. CXCR4 gene sequencing was done detecting a heterozygous p.Arg334STOP mutation, confirming WHIM syndrome. This disease is infrequent and difficult to diagnose.
Key words: immunodeficiency, WHIM, CXCR4, myelokathesis
Recibido: 17-VII-2017 Aceptado: 3-XII-2017
Dirección postal: María Virginia Paolini, Mendoza 2977 4° 13, 1428 Buenos Aires, Argentina
e-mail: virpaolini@gmail.com
El síndrome WHIM (OMIM#193670) es una inmunodeficiencia primaria causada, en la mayoría de los casos, por mutaciones en el receptor de quimiocinas CXCR4 con un patrón de herencia autosómica dominante1-4. El término WHIM es un acrónimo inglés de las principales manifestaciones clínicas del síndrome, verrugas (warts) cutáneo-mucosas de difícil erradicación causadas por el virus del papiloma humano (HPV), hipogammaglobulinemia, infecciones bacterianas recurrentes y mielocatesis, definida por un aumento en médula ósea (MO) de células mieloides maduras asociada a neutropenia en sangre periférica1, 4-7.
Las mutaciones en el extremo carboxi-terminal del receptor de quimiocinas CXCR4 producen una ganancia de función (GOF) en respuesta a su ligando CXCL12 y una mayor actividad quimiotáctica leucocitaria en MO, lo cual podría explicar la mielocatesis8, 9. La predisposición a infecciones por HPV podría relacionarse con una mayor expresión de CXCR4 en queratinocitos y una disminución de células dendríticas, las que presentarían alteraciones de su capacidad migratoria, de su función antiviral mediada por interferón alfa y falta de expresión de la proteína anti-viral MxA9. La hipogammaglobulinemia podría deberse a una alteración en la migración de linfocitos B al centro germinal ganglionar y, como consecuencia, una respuesta inmune secundaria disminuida9.
El tratamiento se basa en el uso de antibióticos profilácticos, gammaglobulina en dosis sustitutiva y factores estimulantes de colonias de granulocitos (G-CSF) o de granulocitos y macrófagos (GM-CSF) en forma cróni- ca1, 4, 9. Se ha informado un caso de trasplante con células madre hematopoyéticas con buena evolución al momento de la publicación10 y se ha planteado el uso de plerixafor (Mozobil®), un antagonista del CXCR4, como una nueva alternativa terapéutica11. La vacuna para HPV (Gardasil®) podría prevenir o retrasar las lesiones por HPV y disminuir su riesgo de transformación maligna12, 13.
Caso clínico
Mujer de 21 años que comenzó a los 10 meses de edad con lesiones purpúricas asociadas a leucopenia que fueron interpretadas como secundarias a infección viral. Durante el siguiente año presentó múltiples infecciones de vías aéreas superiores, una neumonía con derrame pleural, una celulitis de región umbilical, una celulitis de tobillo como complicación de una varicela y fiebre sin foco aparente en varias oportunidades. Durante los cuadros infecciosos se detectaron niveles disminuidos de IgG plasmática (por debajo de 2SD del valor normal) por lo que recibió gammaglobulina endovenosa en los episodios.
A los dos años, por neutropenia crónica, se le realizó una punción aspiración de MO (PAMO) que presentó serie megacariocítica conservada, eritroide ligeramente disminuida (13%) y mieloide aumentada 57% con ligero predominio de elementos inmaduros. Se indicó tratamiento con G-CSF con buena respuesta. Debido a los cuadros infecciosos recurrentes de vía aérea, a los 12 años se le realizó una tomografía axial computarizada de tórax que evidenció bronquiectasias en lóbulo medio derecho y base izquierda con atelectasia en lóbulo medio derecho. Por la neutropenia persistente, menor a 500 células/mm3, se le realizó una PAMO que mostró nuevamente hiperplasia de serie mieloide con elementos en todas las etapas madurativas.
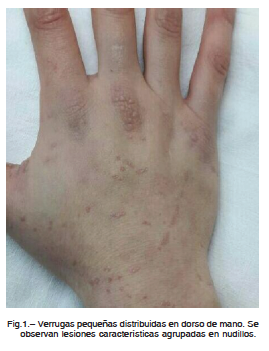
A los 14 años se le diagnosticó herpes zoster en región lumbar. Debido a los múltiples cuadros infecciosos asociados a hipogammaglobulinemia y leucopenia se le realizaron estudios inmunológicos en los cuales presentó niveles disminuidos de IgG, IgM e IgA (por debajo de 2SD de los valores normales), linfopenia con disminución de linfocitos B de memoria (LBm) cuantificados por citometría de flujo (CD19+ – CD27+), respuesta a antígenos proteicos disminuida y a antígenos polisacáridos en el límite inferior de los valores normales, expresión de CD40 en linfocitos B normal y expresión del coestimulador inducible de linfocitos T (ICOS) normal. Se interpretó el cuadro como inmunodeficiencia común variable y comenzó tratamiento sustitutivo con gammaglobulina endovenosa más antibióticos profilácticos con buena respuesta. En el examen funcional respiratorio se diagnosticó un patrón restrictivo leve a moderado. En una ecografía de abdomen se constató esplenomegalia moderada y bazo accesorio en hilio esplénico. A los 20 años se registraron pequeñas verrugas en dorso de ambas manos que progresaron hacia antebrazos, abdomen, cara y rodillas (Fig. 1).
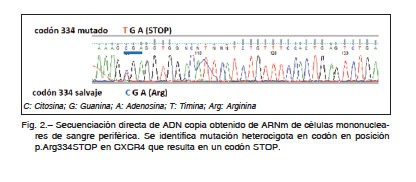
Dado los antecedentes clínicos en el contexto de leucopenia e hipogammaglobulinemia, se decidió realizar estudios moleculares para la búsqueda de mutaciones en el gen CXCR4 en el Área de Biología Molecular del Servicio de Inmunología y Reumatología del Hospital de Pediatría Prof. Dr. Juan P. Garrahan. Por RT-PCR y secuenciación directa se detectó en el gen CXCR4 la presencia de la mutación p.Arg334STOP en estado heterocigota, siendo este resultado compatible con el diagnóstico de síndrome WHIM (Fig. 2)3.
La paciente refirió que su abuelo paterno presentó verrugas durante su vida, su padre y su único hermano no presentaron datos clínicamente relevantes. Actualmente continúa en tratamiento de reemplazo con gammaglobulina subcutánea, G-CSF y azitromicina profiláctica.
Discusión
El síndrome WHIM es una inmunodeficiencia poco frecuente. Si bien el término mielocatesis fue utilizado por primera vez en el año 1964 para describir al cuadro clínico caracterizado por neutropenia crónica asociada a hiperplasia mieloide en MO, no fue hasta 1990 que Wetzler y col. constataron la presencia de verrugas, hipogamma-globulinemia, infecciones bacterianas recurrentes y mielocatesis en varios miembros de una familia y propusieron que estas características formaban parte de una nueva enfermedad congénita a la que denominaron síndrome WHIM1, 14. En el año 2003 Hernández y col. describieron su asociación con mutaciones en el gen CXCR4, molécula inicialmente descripta como correceptor para el virus de la inmunodeficiencia humana3.
Los pacientes presentan una susceptibilidad aumentada a infecciones cutáneo-mucosas por HPV. La edad de aparición de las verrugas varía entre la infancia temprana y la adolescencia tardía4-7. En nuestro caso las lesiones se notaron tardíamente, lo que agregado a lo infrecuente de la enfermedad, hizo que no se sospechara el diagnóstico durante mucho tiempo. Las lesiones papilomatosas que afectan la mucosa genital y oral tienden a diseminarse, son difíciles de erradicar y tienen riesgo de malignización4-7. En este caso, si bien eran leves y no comprometían mucosas, eran extendidas y de difícil resolución. Una mayor susceptibilidad a otras infecciones virales, como al virus varicela-zóster, que presentó nuestra paciente, también han sido comunicadas4-7.
Como en nuestro caso, las infecciones bacterianas recurrentes de vía aérea pueden complicarse con el desarrollo de enfermedad pulmonar crónica, presencia de bronquiectasias pulmonares y exámenes funcionales respiratorios alterados4-7.
La neutropenia, que suele aparecer en forma temprana, puede hacer sospechar una mielodisplasia o una causa autoinmune y quizás, por infrecuente, no evaluar la mielocatesis característica del síndrome WHIM, como ocurrió en este caso.
Los pacientes suelen presentar hipogammaglobulinemia con valores moderadamente o muy disminuidos de IgG asociada a disminución de IgA e IgM y linfopenia B con disminución marcada de los LBm como se registró en este caso1, 4.
Otro factor que causó el retraso diagnóstico fue la sospecha inicial de inmunodeficiencia común variable, causa más frecuente de hipogammaglobulinemia, excluyendo entonces la consideración de este síndrome.
La asociación de verrugas, neutropenia e hipogammaglobulinemia se observan en el 79 al 92% de los pacientes con WHIM, dependiendo de la serie4-7, 15.
A pesar de que las manifestaciones clínicas y de laboratorio resultan bastante típicas, en la práctica el diagnóstico suele verse demorado porque es un síndrome poco frecuente, en el cual la neutropenia podría no ser evidente durante las infecciones, la mielocatesis ser desestimada, el registro de las verrugas tardío y la hipogammaglobulinemia orientar en forma errónea a la inmunodeficiencia común variable.
En otras hipogammaglobulinemias el uso de gamma-globulina en dosis sustitutiva endovenosa o subcutánea ha demostrado ser efectivo en disminuir la frecuencia y gravedad de las infecciones por gérmenes capsulados, por lo que también se indica en pacientes con síndrome WHIM9. En este caso se comenzó en forma endovenosa con buena respuesta y luego se rotó a la forma subcutánea por su eficacia similar y mayor comodidad. El uso de azitromicina profiláctica se basa en la necesidad de reducir la frecuencia y gravedad de las infecciones y por lo tanto la progresión del daño parenquimatoso, estrategia efectiva en otras enfermedades con bronquiectasias e infecciones respiratorias recurrentes. La recomendación del uso de factores estimulantes de colonias de granulocitos, G-CSF o GM-CSF, en forma crónica, podría no ser de utilidad debido a que durante los procesos infecciosos se demostró el aumento de los neutrófilos circulantes9, 15. Por otro lado, no hay estudios concluyentes que demuestren su eficacia en pacientes con WHIM.
El diagnóstico de síndrome WHIM debe sospecharse en pacientes con neutropenia asociada a hipogamma-globulinemia o linfopenia. La ausencia de verrugas no debería excluir el diagnóstico, principalmente en pacientes jóvenes. Se debe investigar la historia familiar y considerar la posibilidad de los casos esporádicos. El diagnóstico de mielocatesis debe ser realizado mediante el estudio de la MO y el de WHIM mediante la secuenciación del gen CXCR4. El tratamiento sustitutivo con gammaglobulina estaría indicado en pacientes con hipogammaglobulinemia e infecciones, y los antibióticos profilácticos en pacientes con enfermedad pulmonar y bronquiectasias. La vacuna contra el HPV podría prevenir, disminuir o evitar la malignización de las lesiones por HPV. La eficacia terapéutica de los factores estimulantes de colonias debería ser demostrada.
Conflicto de intereses: Ninguno para declarar
Bibliografía
1. Wetzler M, Talpaz M, Kleinerman ES, et al. A new familial immunodeficiency disorder characterized by severe neutropenia, a defective marrow release mechanism, and hypogammaglobulinemia. Am J Med 1990; 89: 663-72.
2. Gorlin RJ, Gelb B, Diaz GA, et al. WHIM syndrome, an autosomal dominant disorder: clinical, hematological, and molecular studies. Am J Med Genet 2000; 91: 368-76.
3. Hernandez PA, Gorlin RJ, Lukens JN, et al. Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nat Genet 2003; 34: 70-4.
4. Gulino AV. WHIM syndrome: a genetic disorder of leukocyte trafficking. Curr Opin Allergy Clin Immunol 2003; 3: 443-50.
5. Tassone L, Notarangelo LD, Bonomi V, et al. Clinical and genetic diagnosis of warts, hypogammaglobulinemia, infections, and myelokathexis syndrome in 10 patients. J Allergy Clin Immunol 2009; 123: 1170-3.
6. Dotta L, Tassone L, Badolato R. Clinical and genetic features of warts, hypogammaglobulinemia, infections and myelokathexis (WHIM) syndrome. Curr Mol Med 2011; 11: 317-25.
7. Beaussant Cohen S, Fenneteau O, Plouvier E, et al. Description and outcome of a cohort of 8 patients with WHIM syndrome from the French Severe Chronic Neutropenia Registry. Orphanet J Rare Dis 2012; 7: 71.
8. Bachelerie F. CXCL12/CXCR4-axis dysfunctions: Markers of the rare immunodeficiency disorder WHIM syndrome. Dis Markers 2010; 29: 189-98.
9. Al Ustwani O, Kurzrock R, Wetzler M. Genetics on a WHIM. Br J Haematol 2014; 164: 15-23.
10. Krivanf G, Erdos M, Kallay K, et al. Successful umbilical cord blood stem cell transplantation in a child with WHIM syndrome. Eur J Haematol 2010; 84: 274-5.
11. McDermott DH, Liu Q, Velez D, et al. A phase 1 clinical trial of longterm, low-dose treatment of WHIM syndrome with the CXCR4 antagonist plerixafor. Blood 2014; 123: 2308-16.
12. Handisurya A, Schellenbacher C, Reininger B, et al. A quadrivalent HPV vaccine induces humoral and cellular immune responses in WHIM immunodeficiency syndrome. Vaccine 2010; 28: 4837-41.
13. Rezaei N, Hedayat M, Aghamohammadi A, Nichols KE. Primary immunodeficiency diseases associated with increased susceptibility to viral infections and malignancies. J Allergy Clin Immunol 2011; 127: 1329-41.
14. Zuelzer WW. “Myelokathexis” a new form of chronic granulocytopenia. N Engl J Med 1964; 270: 699-704.
15. Kawai T, Malech HL. WHIM syndrome: congenital immune deficiency disease. Curr Opin Hematol 2009; 16: 20-6.
– – – –
Me explicó que era costumbre yaghan apalear al médico cada vez que un paciente se le muera, como también lo es regalarle cualquier cosa que pida, toda vez que se salve, pues la vida es lo más apreciable que se puede tener.
Fray Mocho (José S. Alvarez) (1858-1903)
En el mar austral (1898), Buenos Aires: Eudeba, 1960, p 118