
ANA B. LATZKE, PEHUÉN FERNÁNDEZ, CARLOS CHIURCHIU, DANIELA SARMANTANO, JAVIER DE ARTEAGA, WALTER DOUTHAT, JORGE DE LA FUENTE
Servicio de Nefrología, Programa de Trasplantes Renales, Hospital Privado Universitario de Córdoba, Universidad Católica de Córdoba, Instituto Universitario de Ciencias Biomédicas de Córdoba, Fundación Nefrológica de Córdoba, Argentina
Resumen El síndrome urémico hemolítico atípico (SUHa) es una entidad rara que se presenta como una
microangiopatía trombótica (anemia hemolítica no inmune, trombocitopenia e insuficiencia renal aguda), cuyas lesiones anatomopatológicas típicas son el engrosamiento de las paredes de capilares y arteriolas con trombosis obstructiva del lumen vascular. Se produce por desregulación de la vía alterna del complemento en la superficie celular, debido a causas genéticas o adquiridas, con una alta tasa de mortalidad, enfermedad renal crónica terminal y recurrencia post-trasplante renal. Las mutaciones de peor pronóstico son las asociadas a factor H, factor B y fracción C3 del complemento. La terapia plasmática resulta útil solo en algunos casos, mientras que el uso de eculizumab es altamente eficaz tanto para el tratamiento agudo como para prevenir las recurrencias en el post-trasplante. Comunicamos el caso de una mujer adulta con diagnóstico de SUHa congénito (mutación de C3) en tratamiento preventivo con eculizumab posterior al trasplante renal, sin recurrencia de la enfermedad, ni efectos adversos relacionados al medicamento a los 36 meses de seguimiento post-trasplante.
Palabras clave: síndrome urémico hemolítico atípico, microangiopatía trombótica, trasplante renal, recurrencia, eculizumab
Abstract Recurrent atypical hemolytic uremic syndrome after renal transplantation: treatment with
eculizumab. Atypical hemolytic uremic syndrome (aHUS) is a rare entity. It is characterized by a thrombotic microangiopathy (nonimmune hemolytic anemia, thrombocytopenia, and acute renal failure), with a typical histopathology of thickening of capillary and arteriolar walls and an obstructive thrombosis of the vascular lumen. The syndrome is produced by a genetic or acquired deregulation of the alternative pathway of the complement system, with high rates of end stage renal disease, post-transplant recurrence, and high mortality. Mutations associated with factor H, factor B and complement C3 show the worst prognosis. Even though plasma therapy is occasionally useful, eculizumab is effective both for treatment and prevention of post-transplant recurrence. We describe here an adult case of congenital aHUS (C3 mutation) under preventive treatment with eculizumab after renal transplantation, with neither disease recurrence nor drug-related adverse events after a 36-months follow-up.
Key words: atypical hemolytic uremic syndrome, thrombotic microangiopathy, renal transplantation, recurrence, eculizumab
Recibido: 22-V-2017 Aceptado: 6-IX-2017
Dirección postal: Ana Belén Latzke, Hospital Privado Universitario de Córdoba, Av. Naciones Unidas 346, 5016 Córdoba, Argentina
e-mail: bellatzke@gmail.com
La microangiopatía trombótica es un síndrome caracterizado por anemia hemolítica microangiopática no inmune, trombocitopenia y fallo renal agudo, cuyas manifestaciones anatomopatológicas son el engrosamiento de las paredes de capilares y arteriolas con trombosis obstructivas del lumen vascular1. Este síndrome puede ser producido por diferentes entidades, siendo las más frecuentes la púrpura trombocitopénica trombótica (o microangiopatía trombótica mediada por déficit de ADAMTS13) y el síndrome urémico hemolítico (SUH) o microangiopatía trombótica mediada por toxina Shiga1.
Una entidad de menor prevalencia es la microangiopatía trombótica mediada por desregulación del complemento, también llamado SUH atípico (SUHa). Este síndrome puede ser hereditario o adquirido, y resulta de una activación descontrolada de la vía alterna del complemento, generando hidrólisis espontánea de C3 a C3b. En ausencia de una regulación normal, el C3b así producido se deposita en los tejidos, generando un incremento en la formación del complejo terminal C5b-9 o complejo de ataque de membrana y lesión de las células afectadas1. La forma hereditaria puede resultar de una mutación de pérdida de función en un gen regulador (CFH, CFI o CD46), o de ganancia de función en un gen efector (CFB o C3) del complemento2, 3. Además, han sido involucrados diferentes polimorfismos. La forma adquirida se produce por la presencia de anticuerpos dirigidos contra componentes reguladores del complemento1.
El SUHa es una entidad agresiva, con mortalidad de 10-15% durante la primera manifestación clínica, progresión a enfermedad renal crónica terminal y recurrencia posterior al trasplante renal en más de 50% de los casos con pérdida del injerto en 80 a 90%4. Las mutaciones de peor pronóstico son las asociadas a CFH, CFB y C35. El tratamiento tradicional consiste en terapia plasmática (intercambio o infusión), con el fin de eliminar las proteínas defectuosas, los anticuerpos contra CFH, y de restaurar las proteínas del complemento de funcionamiento normal. El trasplante hepato-renal también ha sido utilizado en pacientes con mala respuesta al tratamiento. El eculizumab (anticuerpo monoclonal humanizado dirigido contra C5 que inhibe la formación del complejo de ataque de membrana o C5b-9) ha demostrado ser efectivo tanto para el tratamiento previo como el posterior al trasplante, para prevenir la recurrencia post-trasplante, por lo que actualmente es considerado el tratamiento de primera línea6-9. En nuestro país el eculizumab fue utilizado por primera vez en 2013 en un adulto con trasplante renal como tratamiento preventivo de recaída de SHUa.
Caso clínico
Mujer que desarrolló enfermedad renal crónica terminal a los 17 años de edad, secundaria a microangiopatía trombótica. A los dos años de comenzar diálisis recibió su primer trasplante renal de origen cadavérico. Se registró buena función inicial, pero con disminución al desarrollar recurrencia de la microangiopatía trombótica confirmada por biopsia a los 120 días, por lo que debió retornar a hemodiálisis. Dos años después recibió su segundo trasplante con donante vivo relacionado (hermana). A los 14 días post-trasplante presentó microangiopatía trombótica, con hipocomplementemia (C3 bajo) y pérdida abrupta de la función del injerto. Ante la sospecha de SUHa se realizó tratamiento con plasmaféresis sin respuesta favorable y con posterior implantectomía. Permaneció en tratamiento hemodialítico crónico con desaparición de las manifestaciones clínicas y bioquímicas de microangiopatía trombótica, persistiendo siempre niveles bajos de C3.
Nueve años después la paciente consultó en nuestra institución para evaluar la posibilidad de un tercer trasplante renal, por lo que se solicitaron estudios al Instituto Mario Negri (Milán, Italia), siendo evaluada la actividad e inhibición de ADAMTS13 y estudios genéticos del factor B, trombomodulina, factor H, factor I y cofactor proteico de membrana que resultaron normales. El valor de sC5b-9 se encontró elevado: 1436 ng/ml (valor normal: 127-303 ng/ml). Se confirmó la presencia de una mutación heterocigota en el exón 14 del gen que codifica el C3 –C1774T sin sentido determinante de la sustitución de aminoácido Arg592Trp– (también presente en la madre y dos hermanos, uno de ellos donante del segundo trasplante) y cuatro polimorfismos en el mismo gen.
Ante la disponibilidad de eculizumab en nuestro país, la paciente fue inscripta nuevamente en lista de espera para trasplante renal y recibió vacunación anti-meningococo con antelación.
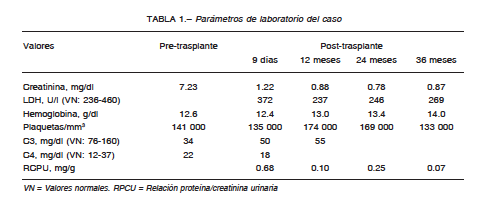
Seis meses después de la inscripción, recibió el tercer trasplante renal, cadavérico, con 12 horas de isquemia fría. Se realizó inducción con plasmaféresis, globulina antitimocítica (1.5 mg/kg), pulsos de esteroides y mantenimiento con tacrolimus (0.2 mg/kg), micofenolato sódico (1440 mg/día) y prednisona. Inició eculizumab en el post operatorio inmediato, 900 mg semanales, y a partir del primer mes 1200 mg cada 14 días. (Fig. 1). Presentó buena evolución clínica y de función del injerto, con alta hospitalaria al noveno día post-trasplante. A los doce días presentó hematuria macroscópica sin causa demostrable, que revirtió espontáneamente. Durante el seguimiento evolucionó con buen estado general, sin complicaciones, con valores en sangre de creatinina sérica, LDH, hemoglobina, plaquetas, C4 y relación proteínas/creatinina en orina estables durante los 36 meses post-trasplante. Los niveles de la fracción C3 del complemento se mantuvieron por debajo de los valores normales (Tabla 1).
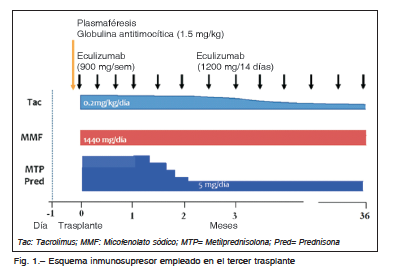
La paciente recibió eculizumab 1200 mg cada 14 días de forma ininterrumpida hasta la fecha, sin presentar infecciones o eventos adversos relacionados con la medicación.
Discusión
Las mutaciones en el C3 representan el 4% de todas las mutaciones asociadas a SUHa. Son de ganancia de función, provocan hiperactivación de la C3-convertasa, llevando a la resistencia de su inactivación, e incrementando la formación del complejo de ataque de membrana en el endotelio10, 11. Están entre las formas de peor pronóstico, con tasas de mortalidad, enfermedad renal crónica terminal y recurrencia del 50-70%10. En nuestro caso, el diagnóstico certero de la enfermedad se realizó luego de la pérdida del segundo injerto, pero es probable que esta enfermedad haya sido también causante del primer episodio de enfermedad renal crónica terminal.
La mayoría de estas mutaciones son heterocigotas, con una penetración del 50%, por lo que es frecuente que existan miembros de la familia como portadores asintomáticos (como en nuestro caso la madre y los hermanos), o con una presentación clínica variable. Esto sugiere que existen factores genéticos y ambientales adicionales (desencadenantes) que modulan el inicio y la progresión de la enfermedad (multiple-hit theory)12.
Existen diferentes mutaciones involucradas en la enfermedad, algunas con peor pronóstico que otras. La caracterización de los componentes genéticos del SUHa, incluyendo la identificación de múltiples mutaciones y polimorfismos en los genes que codifican proteínas del complemento es la clave para predecir la respuesta al tratamiento y la evolución a largo plazo13.
Aunque en la mayoría de las comunicaciones la terapia plasmática no muestra buenos resultados a largo plazo ni elimina las recaídas después del trasplante, la plasmaféresis pre- y post-trasplante reduce la tasa de recidiva de la enfermedad post-trasplante4, 14.
Numerosos estudios demostraron que el eculizumab es efectivo en el tratamiento de SUHa, tanto en riñones nativos como trasplantados, y en la prevención de recaídas post-trasplante6-12. Se recomienda su uso precoz y no se sabe con certeza si, en los pacientes con buena respuesta, se puede interrumpir la administración de este fármaco en algún momento, asunto de importancia por su alto costo. Según expertos, los trasplantados con pérdida de implantes previos no serían buenos candidatos a la interrupción del eculizumab15. A pesar de ser bien tolerado, el mayor riesgo es la predisposición a infecciones, por lo que, al esquema inmunológico estándar, se recomienda agregar la vacuna contra Neisseria meningitidis, la que deberá ser administrada al menos dos semanas antes del medicamento, como se realizó en el caso presentado.
En resumen, el caso comunicado corresponde a un adulto en Argentina que ilustra la eficacia del bloqueo de C5 con eculizumab en la prevención de la recurrencia del SUHa pos-trasplante y el óptimo perfil de seguridad de su administración a mediano plazo.
Conflicto de intereses: Ninguno para declarar
Bibliografía
1. George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med 2014; 371: 654-66.
2. Noris M, Caprioli J, Bresin E, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol 2010; 5: 1844-59.
3. Bu F, Maga T, Meyer NC, et al. Comprehensive genetic analysis of complement and coagulation genes in atypical hemolytic uremic syndrome. J Am Soc Nephrol 2014; 25: 55-64.
4. Noris M, Remuzzi G. Managing and preventing atypical hemolytic uremic síndrome recurrence after kidney transplantation. Curr Opin Nephrol Hypertens 2013; 22: 704-12.
5. Loirat C, Frémeaux-Bacchi V. Atypical hemolytic uremic syndrome. Orphanet J Rare Dis 2011; 6: 60.
6. Zuber J, Le Quintrec M, Krid S, et al. Eculizumab for atypical hemolytic uremic syndrome recurrence in renal transplantation. Am J Transplant 2012; 12: 3337-54.
7. Chatelet V, Fremeaux-Bacchi V, Lobbedez T, Ficheux M, Hurault de Ligny B. Safety and long-term efficacy of eculizumab in a renal transplant patient with recurrent atypical hemolytic-uremic syndrome. Am J Transplant 2009; 11: 2644-5.
8. Licht C, Greenbaum L, Muus P. Efficacy and safety of eculizumab in atypical hemolytic uremic syndrome from 2-year extensions of phase 2 studies. Kidney Int 2015; 87: 1061-73.
9. Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med 2013; 368: 2169-81.
10. Noris M, Remuzzi G. Thrombotic Microangiopathy after kidney transplantation. Am J Transplant 2010; 10: 1517-23.
11. Frémeaux-Bacchi V, Miller EC, Liszewski MKl. Mutations in complement C3 predispose to development of atypical hemolytic uremic syndrome. Blood 2008; 112: 4948-52.
12. Caprioli J, Castelletti F, Bucchioni S, et al. Complement factor H mutations and gene polymorphisms in haemolytic uraemic syndrome: the C-257T, the A2089G and the G2881T polymorphisms are strongly associated with the disease. Hum Mol Genet 2003; 12: 3385-95.
13. De Vriese A, Sethi S, Van Praet J, Nath KA, Fervenza FC. Kidney disease caused by dysregulation of the complement alternative pathway: an etiologic approach. J Am Soc Nephrol 2015; 26: 2917-29.
14. Loirat C, Garnier A, Sellier-Leclerc AL, Kwon T. Plasmatherapy in atypical hemolytic uremic syndrome. Semin Thromb Hemost 2010; 36: 673-81.
15. Goodship TH, Cook HT, Fakhouri F, et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int 2017; 91: 539-51.
– – – –
Everything must have a beginning, to speak in Sanchean phrase; and the beginning must be linked to something that went before. The Hindoos give the world an elephant to support it, but they make the elephant stand upon a tortoise. Invention, it must be humbly admitted, does not consist in creating out of void, but out of chaos; the materials must, in the first place, be afforded: it can give form to dark, shapeless substances, but cannot bring into being the substance itself. In all matters of discovery and invention, even of those that appertain to imagination, we are continually reminded of the story of Columbus and his egg. Invention consists in the capacity of seizing on the capabilities of a subject, and in the power of moulding and fashioning ideas suggested to it.
Todo tiene que tener un comienzo, para decirlo en una frase sanchopancesca; y ese comienzo debe estar ligado a algo que ocurrió antes. Los hindúes dieron al mundo un elefante para sostenerlo, pero hicieron que el elefante estuviera parado sobre una tortuga. La invención, debe admitirse humildemente, no consiste en crear del vacío, sino del caos; los materiales deben, en primer lugar, conseguirse: puede dar forma a sustancias oscuras e informes, pero no puede hacerlas de la nada. En todo asunto de descubrimiento e invención, aun en aquellos que pertenecen a la imaginación, continuamente nos recuerdan la historia de Colón y su huevo. La invención consiste en la capacidad de aprovechar las potencialidades de un tema, y en poder moldearlo y modelar las ideas sugeridas por él.
Mary Shelley
(Mary Wollstonecraft Godwin Shelley) (1797-1851)
Prefacio de la edición revisada de Frankenstein de 1831. En: Frankenstein or
the Modern Prometheus (1818). London: Dent, 1933. Introduction, p vii