
MARÍA VICTORIA COLLADO1, MARÍA DE LOS ÁNGELES GARGIULO1, RAMIRO GÓMEZ2, GRACIELA GÓMEZ1, NICOLÁS PÉREZ1, LORENA SUAREZ1, ANA LÍA TARATUTO3, PATRICIA ARUJ4
1Sección Inmunología, Instituto de Investigaciones Médicas Alfredo Lanari, 2Hospital de Clínicas Gral. José de San Martín 3Consultoría en Neuropatología y Patología Neuromuscular, Instituto de Investigaciones Neurológicas-FLENI, Hospital Nacional de Pediatría J.P Garrahan, 4Sección Neumonología, Instituto de Investigaciones Médicas Alfredo Lanari, UBA, Buenos Aires, Argentina
Resumen La dematomiositis es una miopatía inflamatoria idiopática con espectro clínico variable. En los últimos años se ha identificado un número de autoanticuerpos específicos de miositis útiles para el diagnóstico, la clasificación y el pronóstico de las diversas formas de la enfermedad, entre los que se encuentra el anti-MDA5. Este anticuerpo se asocia al desarrollo de úlceras cutáneas, enfermedad intersticial pulmonar rápidamente progresiva, mortalidad temprana y mal pronóstico por lo que la detección del mismo, en un contexto clínico adecuado, plantea la necesidad de un tratamiento inmunosupresor agresivo. Describimos un caso de dermatomiositis hipomiopática, (es decir, con afección muscular leve) que presentaba compromiso cutáneo específico, enfermedad pulmonar intersticial y anticuerpo anti-MDA5 que respondió favorablemente al tratamiento combinado con ciclofosfamida, gamaglobulina y corticoides.
Palabras clave: dermatomiositis, anticuerpos anti-MDA5, autoanticuerpos, enfermedad pulmonar intersticial
Abstract Dermatomyositis associated with anti-MDA5 autoantibody. Dematomyositis is an idiopathic inflammatory myopathy with a variable clinical spectrum. In recent years, a number of myositis-specific antibodies have been identified including anti-MDA5, which is us eful for diagnosis, prognosis and classification of the diverse clinical forms of the disease. This antibody is associated with cutaneous ulcers, rapidly progressive interstitial lung disease, early mortality and poor prognosis, so the detection of this antibody in a suitable clinical context, raises the need for an aggressive immunosuppressive treatment. We describe a case of dermatomyositis classified as hypomyopathic (i.e. involving mild muscle weakness), presenting specific skin lesions, interstitial lung disease, and presence of anti-MDA5 antibody that had a favorable response to combined treatment with cyclophosphamide, gamma globulin and corticosteroids.
Key words: dermatomyositis, melanoma differentiation associated protein-5, autoantibodies, interstitial lung disease
Recibido: 22-XI-2017 Aceptado: 20-III-2018
Dirección postal: María Victoria Collado, Sección Inmunología, Instituto de Investigaciones Médicas Alfredo Lanari, Combatientes de Malvinas 3150, 1427 Buenos Aires, Argentina
e-mail: vicocollado@hotmail.com
La dermatomiositis pertenece al grupo de miopatías inflamatorias idiopáticas. Se caracteriza por debilidad muscular, lesiones cutáneas distintivas e infiltrado inflamatorio con atrofia perifascicular en la biopsia muscular. Puede asociarse a compromiso pulmonar, cáncer y anticuerpos específicos de miositis, lo que determina la evolución y pronóstico de la enfermedad. Presentamos un caso de dermatomiositis hipomiopática asociada al autoanticuerpo anti-MDA5.
Caso clínico
Mujer de 36 años, mestiza1, que comenzó en noviembre de 2016 con lesiones cutáneas eritematosas en región metacarpofalángica, acompañadas de fenómeno de Raynaud, mialgias y disfagia a sólidos. En ese momento presentaba eritrosedimentación elevada (84 mm/h); enzimas musculares: TGO/P 171/163 UI/l, CPK 108 UI/l, LDH 266 UI/l, aldolasa 8.7 mU/ml, serología negativa para HIV; anticuerpo antinuclear (inmunofluorescencia indirecta, Kallestad HEp-2 Cell line slide, Bio-Rad), anti DNA (Enzimo Inmunoensayo, INOVA) y factor reumatoideo (aglutinación de partículas, Artritest directo Maxi Wiener) negativos; complemento normal: C3: 114 mg%, C4: 29 mg%. (Inmunodifusión Radial, Biocientífica). Ecografía abdominal normal.
Concurrió a nuestro Instituto 5 meses después del inicio de los síntomas. Refería disnea CF II-III, disfonía y disfagia a sólidos. Examen físico: peso 60 kg, debilidad muscular simétrica proximal en las cuatro extremidades (MMT8 110/150)2, signo de Gottrón, eritema en V del cuello, eritema periungueal, y rales secos bibasales. En abril de 2017 se solicitaron pruebas de laboratorio: TGO/TGP: 212/156 UI/l, LDH: 403 UI/l, CPK: 64 UI/l, aldolasa 64 mU/ml; laboratorio inmunológico: anti: RNP (A, B, 70k), Sm (B, D), Ro (52/60), La, Scl-70, cenp-B, Jo-1, p-ribosomal, histonas (Inmunoblot. Inno-LIA Ana Update. Fujirebio) y cardiolipinas IgG e IgM (Enzimo Inmuno Ensayo. BioSystems) que fueron negativos; y anticuerpos asociados y específicos de miositis anti-Mi 2, Jo-1, PM-Scl-100, Pl-7, Pl- 12, Ku (p70/80) y SRP (Immunoblot Myositis Plus. Orgentec) que también fueron negativos. El electromiograma de los cuatro miembros resultó normal. La capilaroscopía demostró disminución de número de capilares. En mayo de 2017 se realizó tomografía de tórax de alta resolución: engrosamiento reticular en ambas bases (Fig. 1); funcional respiratorio: CVF (L): 2 (3.47) 57%, VEF1 (L): 1.71 (2.88) 59%, VEF1/CVF (%) 85 (83)102%; caída moderadamente grave de la capacidad vital con DLCO (ml/min/mmHg) con disminución moderada: 11.88 (21.31) 55%. Ecocardiograma doppler y tomografía por emisión de positrones: normal.
Se hace diagnóstico presuntivo de dermatomiositis con compromiso pulmonar. En mayo comienza tratamiento con meprednisona 1 mg/kg/día, hidroxicloroquina 200 mg c/12 horas, nifedipina 20 mg/día, trimetroprima-sulfametoxazol 160/800 mg 3 v/semana, citrato de calcio 1500 mg- vitamina D3 400 UI c/12 h y gastroprotección. Al mes de iniciado el tratamiento hay mejoría de la disfagia pero persistencia de disfonía y la disnea. Evolucionó con lesiones ulceradas bilaterales tipo necróticas en segunda articulación metacarpofalángica por lo que se sospecha dermatomiositis asociada a anticuerpo anti MDA5 y motiva la solicitud de test ampliado de anticuerpos específicos de miositis que incluye anti: Ku, SRP, PM-Scl, Ej, Oj, NXP2, SAE1, Pl-7, Pl-12, TIF-1γ y anti MDA5 (Inmunoblot. Euroline Autoimmune Inflamatory Myophaties 16 Ag, IgG. Euroimmun). Se agrega al tratamiento con corticoides gamaglobulina ev (2 g/kg), con lo que se logra mejoría parcial de las lesiones cutáneas, de la disfonía, así como de la disfagia y disnea.
En agosto de 2017 se repiten estudios de función pulmonar: CVF (L): 2.03 (3.52) 57%, VEF1 (L): 1.67 (2.92) 57% VEF1/CVF (%) 82 (83) 98% (patrón restrictivo moderado grave) con DLCO (ml/min/mmHg): 13.86 (21.84) 63%, PIMAX 52 cm H2O (75%) y PEMAX 84 cm H2O (104%). Para la misma época se reciben los resultados de la biopsia muscular realizada en el vasto lateral izquierdo que mostró extensa atrofia perifascicular, algunas fibras con núcleos internos y fibras basófilas de tipo regenerativo compatible con dermatomiositis (Fig. 2A) y del anticuerpo anti MDA5 cuyo resultado es positivo. Se hace diagnóstico final de dermatomiositis hipomiopática con enfermedad intersticial pulmonar y anticuerpo anti-MDA5. Se considera necesario agregar al tratamiento ciclofosfamida ev mensual (0.75 mg/m2) y continuar con gamaglobulina teniendo en cuenta el mal pronóstico que presentan la enfermedad pulmonar intersticial y el compromiso cutáneo ante la presencia de este anticuerpo. Luego de 2 meses de tratamiento combinado con ciclofosfamida (dosis acumulada 1.2 g), gamaglobulina y corticoides, se constata enzimas musculares normales, mejoría de las lesiones cutáneas y de los síntomas de disnea, disfagia y disfonía. La Fig. 2B y 2C muestran las lesiones cutáneas de mano antes y después del tratamiento.
Discusión
La dematomiositis es una miopatía inflamatoria idiopática con espectro clínico variable. Su incidencia en Argentina es de 3.2/millón habitantes/año según Rosa y col.3. Los pacientes con dermatomiositis pueden presentar distintos tipos de lesiones cutáneas y grados de compromiso muscular. Cuando el compromiso muscular está ausente o es mínimo, la dermatomiositis se clasifica como amiopática o “hipomiopática”4. Esta situación está frecuentemente asociada a la presencia de anti-MDA5. Este caso se presentó
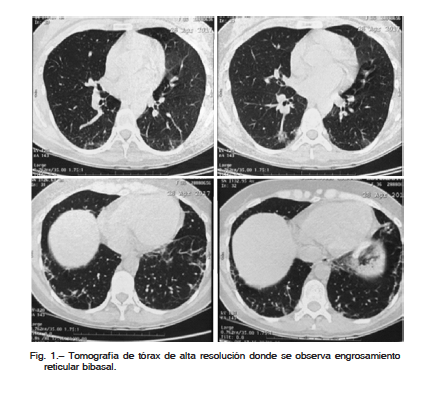
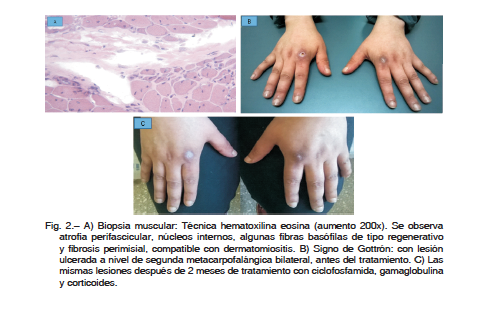
con escasa sintomatología clínica, y enzimas musculares levemente aumentadas. La atrofia perifascicular en la biopsia muscular, característica de esta enfermedad, confirmó el diagnóstico.
La identificación de anticuerpos específicos permite discriminar entre fenotipos clínicos de miositis y valorar posibles complicaciones asociadas como compromiso intersticial pulmonar o cáncer, las que determinarán el pronóstico5. También es útil para hacer diagnóstico diferencial con enfermedades musculares genéticas6. El autoanticuerpo anti-MDA5 (melanoma differentation-associated gene 5), específico de miositis, fue identificado en primera instancia por Sato y col.7 en pacientes japoneses con dermatomiositis amiopática y enfermedad intersticial pulmonar. El autoantígeno reconocido por el anticuerpo anti-CADM140 fue más tarde identificado como un receptor de RNA citoplasmático, codificado por el gen IFIH1. Estaría involucrado en la respuesta inmune innata y se postula que la inducción de interferón I sería el posible mecanismo de autoinmunidad8. CADM140 es idéntico a la proteína 5 asociada a diferenciación de melanoma, por lo que el anticuerpo anti-CADM140 actualmente se denomina anti-MDA5. La prevalencia de anti-MDA5 varía entre el 3 y el 58% de los pacientes con dermatomiositis y aumenta al 100% en los casos de dermatomiositis amiopática9. Se encuentra fuertemente asociado al desarrollo de enfermedad intersticial pulmonar rápidamente progresiva, mortalidad temprana y mal pronóstico. Según un metaanálisis10, su presencia permite identificar el riesgo de desarrollar enfermedad intersticial pulmonar con sensibilidad del 77% y especificidad del 86%, independientemente de la etnia del paciente. La sobrevida a 6 meses en los pacientes con enfermedad intersticial pulmonar rápidamente progresiva y dermatomiositis amiopática se estimó en 41%11. El compromiso rápidamente progresivo se ha observado con mayor frecuencia en la población asiática con respecto a la occidental y por tanto se ha postulado que las características del compromiso pulmonar podrían depender de la etnia12. En el presente caso la paciente es mestiza y su cuadro clínico pareciera similar al de la población occidental.
La pesquisa del compromiso pulmonar con imagen tomográfica no se pone en duda en aquellos pacientes que presentan síntomas. Sin embargo, la evaluación mediante estudios que implican irradiación en pacientes asintomáticos es cuestionable. Esto determina que la frecuencia del compromiso pulmonar varíe según los métodos diagnósticos utilizados. Ello realza la utilidad de métodos menos riesgosos, tales como la detección del anticuerpo anti-MDA5, ante la sospecha clínica de complicación pulmonar. En el presente caso, la paciente presentaba disnea y lesiones cutáneas ulceradas, lo cual implicó la realización de estudio de función pulmonar, tomografía computarizada de tórax de alta resolución y determinación de anti-MDA5. El resultado positivo del anticuerpo anti-MDA5 apoyó la sospecha clínica y el compromiso pulmonar se evidenció en los estudios funcionales y en la tomografía. Dado su carácter rápidamente progresivo y alta mortalidad, el compromiso pulmonar requiere tratamiento inmunosupresor severo y de inicio precoz. Por tal motivo, en este caso se inició tratamiento con ciclofosfamida ev.
Por otro lado, la presencia de anti-MDA5 también se asocia a la aparición de úlceras cutáneas, úlceras digitales y manos edematizadas, debido a vasculopatía grave y de difícil tratamiento13. En estas circunstancias, una opción terapéutica es la de inmunoglobulina. Este caso refractario al tratamiento corticoideo y vasodilatador con nifedipina, mejoró tras la aplicación de gamaglobulina endovenosa.
Una vez iniciado el tratamiento, el descenso del título del anticuerpo se postula como un marcador de respuesta efectiva al mismo, por lo cual sería de utilidad la determinación repetida del mismo para el seguimiento14. En el presente caso no ha sido factible medir los títulos del anticuerpo ya que la técnica disponible es cualitativa.
Los pacientes con dermatomiositis amiopática presentan menor riesgo de desarrollar cáncer con respecto a la dermatomiositis clásica. Sin embargo, algunos autores recomiendan la búsqueda de neoplasia oculta, con frecuencia anual durante al menos los dos primeros años desde el diagnóstico3. La presencia de enfermedad intersticial pulmonar tiene asociación negativa con el desarrollo de cáncer, aunque hacen falta más estudios para determinar por qué éste podría ser un mecanismo protector. La solicitud de PET en este caso se realizó con la finalidad de descartar esta posibilidad y teniendo en cuenta que este tipo de estudio es costo-efectivo en comparación a la pesquisa clásica de cáncer acorde a sexo y edad15. Se descartó así la existencia de neoplasia oculta.
En conclusión, en nuestro conocimiento, este es el primer caso publicado de dermatomiositis asociado al anticuerpo anti-MDA5 en Argentina. Las lesiones cutáneas, el escaso compromiso muscular y la disnea orientaron a solicitar la determinación del anticuerpo anti-MDA5, característico en esta entidad. La presencia de dicho anticuerpo y los estudios pulmonares permitieron realizar un diagnóstico precoz, prever la evolución clínica e iniciar tratamiento inmunosupresor temprano y severo, sobre todo para reducir el compromiso pulmonar, que es el factor pronóstico más importante en estos casos.
Agradecimientos: La detección de autoanticuerpos asociados a miositis se realizó gracias al aporte económico de la Sociedad Argentina de Reumatología para la compra de kit Euroimmun que gestionó el Grupo de Estudio de Miopatías de la mencionada Sociedad.
Conflicto de intereses: Ninguno para declarar
Bibliografía
1. Pons-Estel BA, Catoggio LJ, Cardiel MH, et al. The GLADEL multinational Latin American prospective inception cohort of 1,214 patients with systemic lupus erythemato sus: ethnic and disease heterogeneity among “Hispanics”. Medicine (Baltimore) 2004; 83: 1-17.
2. Rider LG, Koziol D, Giannini EH, et al. Validation of manual muscle testing and a subset of eight muscles (MMT8) for adult and juvenile idiopathic inflammatory myopathies. Arthritis Care Res (Hoboken) 2010; 62: 465-72.
3. Rosa J, Garrot LF, Navarta DA, et al. Incidence and prevalence of polymyositis and dermatomyositis in a health management organization in Buenos Aires. J Clin Rheumatol 2013; 19: 303-7.
4. Sontheimer RD. MDA5 autoantibody – another indicator of clinical diversity in dermatomyositis. Ann Transl Med 2017; 5: 160.
5. Fujimoto M, Watanabe R, Ishitsuka Y, Okiyama N. Recent advances in dermatomyositis-specific autoantibodies. Curr Opin Rheumatol 2016; 28: 636-44.
6. Mammen AL,Casciola- Rosen L, Christopher-Stine L, Lloyd TE, Wagner RK. Myositis-specific autoantibodies are specific for myositis compared to genetic muscle disease. Neurol Neuroimmunol Neuroinflamm 2015; 2:e172
7. Sato S, Hirakata M, Kuwana M, et al. Autoantibodies to a 140-kd polypeptide, CADM-140, in Japanese patients with clinically amyopathic dermatomyositis. Arthritis Rheum 2005; 52: 1571-6.
8. Sadler AJ. The role of MDA5 in the development of autoimmune disease. J Leukoc Biol 2017; pii: jlb.4MR0617- 223R.
9. Satoh M, Tanaka S, Ceribelli A, Calise SJ, Edward KL, Chan A. Comprehensive overview on myositis-specific antibodies: new and old biomarkers in idiopathic inflammatory myopathy. Clin Rev Allergy Immunol 2017; 52: 1-19.
10. Chen Z, Cao M, Plana MN, et al. Utility of anti-melanoma differentiation-associated gene 5 antibody measurement in identifying patients with dermatomyositis and a high risk for developing rapidly progressive interstitial lung disease: a review of the literature and a meta-analysis. Arthritis Care Res (Hoboken) 2013; 65: 1316-24.
11. Ye S, Chen XX, Lu XY, et al. Adult clinically amyopathic dermatomyositis with rapid progressive interstitial lung disease: a retrospective cohort study. Clin Rheumatol 2007; 26: 1647-54.
12. Chen Z, Wang Y, Kuwana M. HLA-DRB1 alleles as genetic risk factors for the development of anti-MDA5 antibodies in patients with dermatomyositis. J Rheumatol 2017; 44: 1389-93.
13. Fiorentino D, Chung L, Zwerner J, Rosen A, Casciola- Rosen L. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol 2011; 65: 25-34.
14. Sato S, Kuwana M, Fujita T, Suzuki Y. Anti-CADM-140/ MDA5 autoantibody titer correlates with disease activity and predicts disease outcome in patients with dermatomyositis and rapidly progressive interstitial lung disease. Mod Rheumatol 2013; 23: 496-502.
15. Selva-O’Callaghan A, Grau JM, Gámez-Cenzano C, et al. Conventional cancer screening versus PET/CT in dermatomyositis/polymyositis. Am J Med 2010; 123: 558-62.