
VÍCTOR A. GAONA
Servicio de Neuropediatría, Centro Médico La Costa, Asunción, Paraguay
Resumen En un amplio espectro de casos en la infancia, la macrocefalia no conlleva un riesgo neurológico
grave, aunque un abanico de posibles afecciones tendrá impacto significativo, tanto en los aspectos evolutivos como cognitivos de los niños. Lo anterior ocurre en enfermedades con componentes progresivos, como tumores o hidrocefalia, y en aquellos casos en los cuales el factor del crecimiento del perímetro cefálico está dado por componentes estructurales del sistema nervioso como sucede en las megalencefalias. Como en todos los demás actos médicos, la toma cuidadosa de la anamnesis, el adecuado examen neurológico y las valoraciones de los ítems del neurodesarrollo podrán dar una orientación cabal acerca de la etiología e importancia del problema. La ayuda de los medios auxiliares de diagnóstico, así como los de imágenes, proveerán de los demás datos para definir el diagnóstico y plantear un tratamiento.
Palabras clave: macrocefalia, megalencefalia, hidrocefalia, neurodesarrollo
Abstract Macrocephaly in childhood. In a wide spectrum of cases in childhood, macrocephaly does not carry
a neurological risk, although a range of possibilities will have an impact on both the evolutionary and cognitive aspects of children. The previous happens in pathologies with progressive components, such as tumors or hydrocephalus, and in those cases in which the factor of the growth of the cephalic perimeter is given by structural components of the nervous system as it happens in megalocephaly. As in all other medical acts, the careful taking of the anamnesis, the appropriate neurological examination and the valuations of the neurodevelopment items can give a thorough orientation about the etiology and importance of the problem. The help of diagnostic aids as well as images will provide the other data to define the diagnosis and propose a treatment.
Key words: macrocephaly, megaloencephaly, hydrocephalus, neurodevelopment
e-mail: vgaona@gmail.com
Se entiende como macrocefalia al del perímetro craneal (PC) más allá de dos desviaciones estándar (+2DE) por encima del percentil 95 establecidos para edad, sexo y etnia, y su prevalencia es de aproximadamente un 5% de la población general1. El desarrollo del PC está condicionado por el crecimiento de sus componentes: el líquido cefalorraquídeo (LCR), el volumen sanguíneo y la masa estructural cerebral; un crecimiento superior en cualquiera de estos elementos podrá resultar en un mayor tamaño craneano.
En el último trimestre del embarazo y los dos primeros años de vida se produce el mayor incremento del PC, esta variación está en relación al propio desarrollo del sistema nervioso central (SNC) y se constituye en una manera indirecta de seguir la evolución del SNC como parte importante del protocolo de evaluación pediátrica.
En términos generales, en un niño nacido a término, se tiene un perímetro cefálico de unos 35 cm. Es la primera medición que se realiza del PC, medida que luego será utilizada para controlar el incremento de este PC conforme a las tablas estandarizadas de seguimiento antropométrico. Se debe considerar que algunas mediciones pueden ser afectadas y variar, sobre todo en el recién nacido (RN), debido a situaciones particulares como la presencia de cabalgamiento óseo, cefalohematomas, colecciones líquidas o serosanguíneas y otras.
El crecimiento del PC sigue por regla general un desarrollo de 2 cm por mes (en el primer trimestre de vida), 1 cm por mes (en el segundo trimestre de vida) y unos 0.5 cm por mes (en los siguientes dos trimestres de vida); completando así un crecimiento de unos 12 cm en el primer año de vida. Posteriormente, se agregan unos 0.5 cm por mes hasta la edad de dos años, completando así el período de mayor crecimiento y expansión del sistema nervioso.
Es interesante definir macrocefalia y megalencefalia, utilizados indistintamente como similares o sinónimos. Si bien ambas utilizan el parámetro de +2DE por encima de lo esperado para edad y sexo, la megalencefalia es un término que se refiere al crecimiento del PC ocasionado por un mayor desarrollo de los componentes estructurales del cerebro; ya sea por proliferación neuronal, trastornos en la migración neuronal o como consecuencias post natales que generen incrementos de los componentes del sistema nervioso central y que no se acompaña generalmente del aumento de la presión intracraneana. La macrocefalia, se refiere al incremento del PC generado por una amplia variedad de causas, tales como colecciones de LCR o hidrocefalias comunicantes o no, masas proliferativas u ocupativas de espacio, alteraciones óseas o malformaciones vasculares, entre otras2.
Clasificación
Diversos autores clasifican las macrocefalias conforme a múltiples variables, tales como su origen (adquiridas o congénitas), su evolución (estáticas o progresivas), acompañadas o no de hipertensión intracraneana o conforme a cuál es el elemento que genera el incremento del PC; sea este el LCR, el componente sanguíneo, la propia estructura cerebral o el sistema vascular.
Una forma simple de clasificarlas es la siguiente:
Macrocefalias asociadas a enfermedad cerebral o del LCR
– Primarias: con aumento del tamaño y peso del cerebro
– Constitucionales
– Hemimegalencefalia
– Síndromes genéticos
– Secundarias: con ocupación o desplazamiento de estructuras
– Lesiones ocupantes de espacio
– Hidrocefalias incluyendo la externa benigna de la infancia
– Depósitos de sustancias
Macrocefalias asociadas a afecciones óseas
– Craneana por alteración en los procesos de cierre de suturas
– Dolencias óseas sistémicas
Sintomatología
La macrocefalia no es una enfermedad en sí misma, sino que representa la manifestación de un problema subyacente, por ende, la manifestación neurológica tenderá a representar más a la enfermedad de base que a la misma macrocefalia. El paciente se puede presentar:
Asintomático
-En la macrocefalia constitucional o en la hidrocefalia externa benigna
Sintomático
– Procesos ocupativos de espacio e hidrocefalias: retardo del desarrollo (RD), irritabilidad, cefaleas, vómitos, déficits neurológicos y crisis convulsivas
– Síndromes genéticos o alteraciones por depósitos de sustancias: deterioro neurológico y/o cognitivo y crisis convulsivas. Rasgos dismórficos y fenotipo particular, hepato-esplenomegalia, estigmas cutáneos
– Hemimegalencefalia: epilepsia, RD, hemiparesia
– Alteraciones en el cierre de suturas o craneosinostosis: alteración en la forma craneana, síndrome de hipertensión intracraneana, déficits neurológicos, sobre todo visuales
– Enfermedades óseas sistémicas: alteración del medio interno o bioquímico, retraso pondoestatural, deformidad esquelética y compromiso general
Anamnesis y examen
Como en todas las enfermedades, una cuidadosa revisión y toma de antecedentes es vital para el enfoque diagnóstico y establecer los diagnósticos diferenciales. Se debe interrogar y valorar los antecedentes pre y perinatales tales como exposición a radiaciones, drogas o tóxicos. La posibilidad de infecciones maternas, partos complicados o traumáticos y sufrimiento fetal, considerando los controles de ecografías y/o monitoreos gineco-obstétricos que ya pueden orientar pre natalmente hacia el diagnóstico.
No olvidar preguntar acerca de problemas similares o neurológicos en las familias de los progenitores y detallando ítems que conforman el neurodesarrollo normal para edad del paciente.
Poner particular atención a:
– El perímetro y forma del cráneo, con mediciones extrapoladas a una curva de crecimiento estándar para detectar variaciones, anticipando efectos de las hidrocefalias progresivas o procesos expansivos. Examinar la fontanela que no debe ser protruyente y variar con los ciclos respiratorios. Auscultar el cráneo en busca de soplos y palpar las suturas craneanas.
– Valorar dismorfias o alteraciones fenotípicas que orienten hacia enfermedades genéticas, examinar la piel en busca de estigmas neurocutáneos. Buscar e interrogar sobre aromas de piel u orina en enfermedades metabólicas. Detectar hepatoesplenomegalia.
– Examinar tono, fuerza y reflejos. Pares craneales, déficits sensoriales, desarrollo psicomotor, coordinación motora, presencia de signos meníngeos, etc.
Métodos auxiliares de diagnóstico
Son utilizados para aseverar el diagnóstico, detallar anomalías asociadas, prever complicaciones y establecer un plan terapéutico. Su premura la define la velocidad del incremento del PC, diferenciando entre evolutivas y no evolutivas, siendo las primeras las que requieren prioritariamente estudios de control. Otros parámetros son los signos asociados de hipertensión endocraneana, anomalías neurocutáneas, historia previa de traumatismos o infecciones del SNC, anomalías dismórficas, historia familiar previa o afectación del neurodesarrollo.
– Ecografía transfontanelar: Siempre que se tenga un acceso adecuado por medio de las fontanelas, este método es muy valorado por la rapidez con que se lo puede realizar, la inocuidad del método que no requiere de sedación o preparativos previos, la facilidad de hacer un seguimiento por medio de estudios seriados, el fácil acceso al medio hospitalario y su bajo costo. Se pueden apreciar sobre todo dilataciones ventriculares, desvíos de línea media, colecciones o masas ocupativas, entre otras.
– Tomografía de cerebro: Agrega a lo anterior la posibilidad de valorar el sistema vascular por medio del uso de contrastes y la visualización de calcificaciones o masas, captadoras o no de contraste, en procesos proliferativos. Con el uso de la reconstrucción en 3D se valora la forma del cráneo y el estado de cierre de las suturas en los casos de craneosinostosis.
– Resonancia magnética cerebral: Es el estudio de elección. Agrega una visión excelente del sistema nervioso valorando adecuadamente procesos malformativos, degenerativos, trastornos de la migración neuronal, hidrocefalias activas o estacionarias, colecciones o quistes y procesos tumorales. Con el agregado de la tractografía podemos explorar los trayectos de los haces nerviosos y con la espectroscopia determinar la posible naturaleza bioquímica de la zona estudiada. Adicionando angiografía apreciamos el árbol vascular arterial y venoso.
– Otros estudios: Se pueden solicitar estudios genéticos para cromosomopatías o genopatías, estudios químicos o enzimáticos en las etiologías metabólicas, EEG para considerar comicialidad asociada, electromiografía y VCM en neuropatías periféricas, fondo de ojo en procura de anomalías retinianas, ecografías abdominales en enfermedades de depósito y evaluación dermatológica para lesiones en piel asociadas a anomalías del SN.
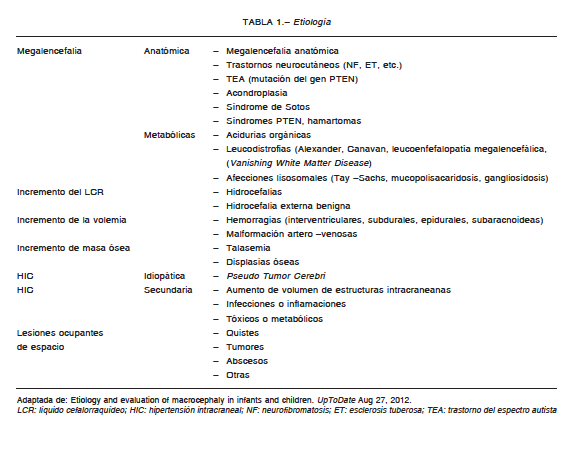
Etiología
Es variada y amplia, enfatizaremos algunas entidades que nos parecen interesantes por sus características e importancia (Tabla 1)
Hidrocefalia
Es la acumulación del LCR ocasionando aumento del volumen ventricular y generando desplazamiento de estructuras cerebrales con consecuencias deletéreas sobre el mismo; su pronta detección y corrección inciden favorablemente en el pronóstico. Normalmente existe un equilibrio entre la producción, circulación y la reabsorción del LCR asegurando presión y volumen intracraneano estable; la ruptura de este equilibrio desembocará en hidrocefalia y dará origen a los dos tipos clásicos:
Hidrocefalia comunicante
– Dificultades en la reabsorción: Localizadas en las vellosidades aracnoideas que se ocupan de esa tarea afectadas por procesos hemorrágicos, infecciosos, bloqueos de los senos venosos y procesos tumorales locales o metastásicos.
– Aumento de la producción: Como el papiloma de los plexos coroideos, que ocasiona un aumento de la producción del LCR no compensado por los mecanismos de reabsorción.
– Hidrocefalia externa benigna: Es una forma de presentación bastante común en la infancia (5%) de curso benigno y autolimitado, resolución hacia los dos años de vida, sin secuelas de importancia y con marcados antecedentes familiares de macrocefalia. Se estima que su desarrollo se debe a la incapacidad para reabsorber el LCR en la convexidad cerebral, a una desproporción cráneo encefálico o una variable del desarrollo normal del cerebro3.
– Hidranencefalia: Existe ausencia de hemisferios cerebrales con preservación del cerebelo, pedúnculos cerebrales y tálamos. Está ocasionada por infartos antenatales en el territorio de las carótidas4. Se considera que las causas infecciosas se incrementarán debido a, por ejemplo, el brote del Zika, como lo informaron Sarno y col.5 La supervivencia de estos pacientes es pobre y la mayoría fallece en los primeros años de vida, siendo el tratamiento meramente paliativo.
Hidrocefalia no comunicante
Obstrucción al flujo y circulación adecuada del LCR ocasionando acumulación intracraneana del mismo. Se la considera como la más frecuente causa de hidrocefalia de inicio temprano, siendo sus etiologías habituales:
– Estenosis del acueducto de Silvio: Dado su reducido diámetro, es presa fácil de complicaciones como obstrucción por procesos hemorrágicos, tan frecuentes en etapa neonatal, o tumores. Existe una forma genética ligada al cromosoma X por mutación del gen L1CAM (neural cell adhesion molecule L1 gene) a nivel Xq28 con una tasa de recurrencia del 50 %, asociada frecuentemente a otras anomalías como retraso del desarrollo psicomotor grave, afectación de vías piramidales y espasticidad6.
– Síndrome de Arnold-Chiari: Se debe al desplazamiento de las amígdalas cerebelosas y parte del tronco encefálico a través del agujero occipital ocasionando una obstrucción al flujo del LCR. Puede asociarse a meningocele, mielomeningocele, síndrome de médula anclada o encefalocele.
– Síndrome de Klippel-Feil: Es la fusión de vértebras cervicales que ocasiona dificultades al drenaje de IV ventrículo. Presenta pelo de implantación baja, motilidad cervical alterada y movimientos en espejo de manos con fácil diagnóstico con estudios de imágenes.
– Síndrome de Dandy-Walker: Combinación de agenesia del vermis cerebeloso, IV ventrículo quístico y megacisterna magna. Existe obstrucción de los agujeros de Luschka y Magendie con diagnóstico antenatal accesible por ecografía.
– Masas ocupantes de espacio: Entre estos tenemos a los procesos tumorales, con distintas líneas celulares (astrocitomas, gliomas, meduloblastomas, etc.), así como colecciones por hemorragias e infecciones como abscesos o hematomas. Suelen ocasionar aumento del PC de agudo o subagudo y su diagnóstico reposa en las imágenes de TAC o RMN.
– Acondroplasia: Afección autosómico dominante con mutación del gen que codifica el receptor 3 (FGFR3) en el cromosoma 4. Anualmente afecta a uno de cada 26 000 nacimientos7. Puede existir hidrocefalia por desproporción entre la base del cráneo y el cerebro, ocasionando dificultad al drenaje del LCR con estenosis del seno sigmoide.
Megalencefalia
Es el aumento del tamaño o peso del cerebro acompañado o no de alteraciones neurológicas afectando la totalidad o parte del mismo. No se asocian signos de aumento en la presión intracraneana, incrementos del volumen del LCR o presencia de masas ocupantes de espacio. Se ha informado el rol del incremento del número de astrocitos debido al papel de las proteínas CD81 o al factor de crecimiento IGF-1 sobre el número de neuronas8. Clásicamente se clasifica, según etiología, en: benignas o idiopáticas, anatómicas y metabólicas.
Megalencefalia benigna
Se refiere a aquellas en las cuales existe un aumento del perímetro cefálico por encima de lo normal sin alteración en el neurodesarrollo, estacionándose el incremento del tamaño craneal en la adolescencia. Existe una historia familiar de macrocefalia en los padres, por lo que es importante también medir el perímetro cefálico de ellos.
Megalencefalia metabólica
La manifestación está en estrecha relación a la enfermedad de base y la búsqueda del patrón genético (asociación al X, perfil autosómico o recesivo o consanguinidad). Se debe examinar el posible compromiso de otros órganos o sistemas como piel, hepatoesplenomegalia, ojos, nervios y músculos; fallas en el neurodesarrollo o regresión en los ítems ya logrados, así como presencia de crisis convulsivas, hipo e hipertonía, letargia o irritabilidad. Los estudios de laboratorio son de vital importancia en la búsqueda del defecto químico o enzimático que debe ser buscado en suero, orina, LCR o cultivos tisulares.
Existen tres grupos asociados al problema: los defectos de ácidos orgánicos, las leucoencefalopatías metabólicas y las enfermedades por depósito de substancias.
Defectos de ácidos orgánicos
Aciduria glutárica Tipo 1
Existe deficiencia de glutaril CoA deshidrogenasa que participa en la transformación de lisina a hidroxilisina y al triptófano; se acumula ácido glutárico y 3-hidroxiglutárico alterando el metabolismo energético provocando falla neuronal. Se presentan movimientos distónicos, infecciones recurrentes y lesiones en RMN con cisura de Silvio amplia (en alas de murciélago) e hiperintensidad en ganglios basales9.
Aciduria L-2-hidroxiglutárica
Es la más frecuente y se manifiesta con gran falla neurológica y crisis epilépticas. La RMN revela compromiso de sustancia blanca, preferentemente frontal, con hallazgos neuropatológicos de gliosis, pérdida neuronal, desmielinización e hiperplasia astrocitaria10.
Encefalopatías metabólicas
Enfermedad de Alexander
Es una macrocefalia con deterioro progresivo del neurodesarrollo. La RMN demuestra lesiones frontales bilaterales y escasa substancia blanca témporo-occipital. Se afectan primordialmente los astrocitos por una mutación del gen GFAP que codifica la proteína fibrilar de la glía11.
Enfermedad de Canavan
Presente rápido incremento en el PC desde los primeros meses con grave hipertonía y convulsiones, llevando al deceso en la primera década. Se produce por acumulación de ácido N-metilaspartato (NAA) y se verifica degeneración de la sustancia blanca en RMN con eliminación urinaria elevada de NAA12.
Leucoencefalopatía megalencefálica con quistes subcorticales
La macrocefalia puede estar presente desde el nacimiento al primer año de vida, acompañada de crisis epilépticas, hipotonía y retraso del desarrollo psicomotor. La RMN demuestra la afectación de substancia blanca y la presencia de los quistes subcorticales13.
Leucoencefalopatía con desaparición de sustancia blanca
Existe pérdida progresiva de la sustancia blanca con preservación de la gris, constatable en los estudios de RMN. Aparece en los primeros años con afectación motora grave y espasticidad14.
Enfermedades por depósito
Están asociadas a desarrollo de macrocefalia por incremento de sustancias de depósito, tal como la enfermedad de Tay-Sachs, que se presenta con hipotonía y retraso psicomotor en el primer año de vida con incremento del peso cerebral, hasta el doble de lo normal, por depósito de gangliósidos asociados a espasticidad y epilepsia con la característica mancha rojo cereza en examen de fondo de ojo. La enfermedad de Sandhoff, considerada como una variante de la anterior, en la cual también existe depósito de gangliósidos en elevada proporción generando cuadros muy similares15.
Megalencefalia asociada a síndromes genéticos o cromosómicos
Síndrome de Sotos
Afectación del cromosoma 5q35 con macrocefalia, epilepsia, retraso psicomotor y facies características que a 1/10 000 a 30 000 RN. Tienen una mayor incidencia de neoplasias, cardiopatías congénitas y defectos oculares. En los estudios de imágenes se pueden detectar múltiples anormalidades: dilatación ventricular, disgenesias cerebrales, afectación de substancia blanca y atrofia cerebral16.
Síndrome de Weaver
Se solapa mucho con el anterior si bien pueden agregarse llanto ronco, camptodactilia, alteraciones en piel y hernia umbilical.
Síndrome Bannayan-Riley-Ruvalcalba
Con defectos en el gen PTEN, como en el síndrome de Cowden, en el locus 10q23.31 que asocia un sobrecrecimiento del tejido conectivo, múltiples hamartomas y macrocefalia, aumento de talla y peso con tendencia fácil a neoplasias17.
Síndrome de Simpson-Golabi-Behmal
Tiene el defecto localizado en el locus Xq26 con dismorfias faciales y macrocefalia. Anormalidades asociadas en riñones, tracto gastrointestinal, corazón y sistema óseo18.
Síndrome de Pretzel
Asocia polihidramnios, megalencefalia y epilepsia sintomática. Inicia en la infancia por deleción de los exones 9 y 13 del gen LYK5 que controla el crecimiento y desarrollo de varias líneas celulares19. El RD es grave, con inicio temprano de convulsiones y atrofia muscular, adoptando el niño una posición característica similar al alimento del mismo nombre. La RMN y anatomía patológica muestran presencia de hidrocefalia con crecimiento cerebral aumentado y ventrículomegalia acompañada de heteropías subcorticales y subependimarias20.
Macrocefalia y autismo
Es interesante ver la relación que se demuestra entre las mutaciones del gen PTEN, ligado al cromosoma X, en un grupo de pacientes con presencia de trastornos del espectro autista, problemas conductuales y cognitivos con marcada macrocefalia21. Otros trabajos refieren la participación de la mutación RAB1q asociando macrocefalia y autismo22.
Síndrome de Optiz-Kaveggia o síndrome FG
Pacientes con macrocefalia y con retraso psicomotor moderado a grave, ano imperforado, pulgares anchos y planos; dando sensación de aplastamiento, acompañados de dismorfias faciales. El síndrome está generado por una mutación del gen MED12 localizado en el Xq13, gen relacionado también a los síndromes de Ohdo y Luján23, 24.
Síndrome del cromosoma X frágil
Es una afección frecuente que explica hasta 1/3 de las alteraciones cognitivas y conductuales en pediatría. Está ligado al cromosoma X por una mutación en Xq27.3, con expansión anómala de la citosina-guanina-guanina (CGG) en el gen FMR1 (Fragile X Mental Retardation 1), y una incidencia de aproximadamente 1:4000 a 1:6000 recién nacidos. Los pacientes presentan una facies característica con cara alargada, orejas amplias, macrorquidia, alteraciones esqueléticas con hiperlaxitud de ligamentos y alteraciones del SNC25,26.
Neurofibromatosis Tipo 1
Conjunto heterogéneo de lesiones del sistema nervioso central y periférico con patrón de herencia autosómico dominante. La macrocefalia está presente en un elevado número de casos siendo del 20 a 30% de los afectados27. Está causada por una mutación de la neurofibromina en el gen 17q11.2 que controla el sistema de señalización del factor de crecimiento mTOR28. Los estudios de RM en estos pacientes señalan que la macrocefalia está ocasionada principalmente por un incremento en el volumen de la sustancia blanca, sobre todo frontal y parietal, si bien puede coexistir alteración en la sustancia gris29,30.
Tratamiento
El tratamiento variará en función a la enfermedad de base que está generando el problema, y oscilará desde solo un proceso de control, como en la hidrocefalia externa benigna de la infancia que solo requiere de seguimiento dada su evolución favorable, a las hidrocefalias progresivas o procesos ocupativos que serán de resorte quirúrgico difiriendo la técnica y el abordaje para cada caso en particular. En otras situaciones se requieren de terapias nutricionales, enzimáticas o trasplante de médula ósea para enfrentar enfermedades neurometabólicas o de depósito. Como hemos visto, las macrocefalias se presentan generalmente con muchas comorbilidades asociadas. Ello obliga a un enfoque más amplio con aplicación de tratamientos multimodales, conforme al área o función afectada, como fisioterapia, rehabilitación, terapias cognitivas o conductuales, educación especial y adecuación curricular, además de medicación en casos de crisis asociadas o trastornos de conducta. No olvidar el apoyo emocional a los padres y entorno familiar y el consejo genético según el caso.
En conclusión, la macrocefalia es una entidad frecuente en la consulta, sobre todo en la neuropediátrica, y su impacto sobre el niño fluctúa desde lo casi imperceptible hasta consecuencias devastadoras, tanto en el aspecto del desarrollo psicomotor como en el funcional o vital. El correcto seguimiento del desarrollo antropométrico del paciente, la evaluación cuidadosa de su neurodesarrollo y otros aspectos de la práctica médica, serán el disparador de la sospecha y posterior investigación de esta enfermedad que, gracias a la amplia gama de medios de apoyo, proporcionará una inestimable ayuda para poder definir el diagnóstico y establecer la mejor opción terapéutica y orientación a los padres.
Conflicto de intereses: Ninguno para declarar
Bibliografía
1. Hamill PV, Drizd TA, Johnson CL, Reed RB, Roche AF, Moore WM. Physical growth: National Center for Health Statistics Percentiles. Am J Clin Nutr 1979; 32: 607-29.
2. Winden KD, Yuskaitis CJ, Poduri A. Megalencephaly and macrocephaly. Semin Neurol 2015; 35: 277-87.
3. Medina LS, Frawley K, Zurakowski D, Buttros D, De Grauw A, Crone KR. Children with macrocrania: Clinical and imaging predictors of disorders requiring surgery. Am J Neuroradiol 2001; 22: 564-70.
4. Pavone P, Practicò AD, Vitaliti G, Ruggieri M, Rizzo R, Parano E. Hydranencephaly: cerebral spinal fluid instead of cerebral mantles. Ital J Pediatr 2014; 40: 79.
5. Sarno M, Sacramento GA, Khouri R, et al. Zika virus infection and stillbirths: a case of hydrops fetalis, hydranencephaly and fetal demise. PLoS Negl Trop Dis 2016; 10: e0004517.
6. Santos F, Temudo T. Hidrocefalia ligada al cromosoma X (síndrome de Bickers-Adams). Presentación de un caso confirmado por la genética molecular. Rev Neurol 2000; 31: 1039-42.
7. Bagley CA, Pindrik JA, Bookland MJ, Camara-Quintana JQ, Carson BS. Cervicomedullary decompression for foramen magnum stenosis in achondroplasia. J Neurosurg 2006; 104 (3 suppl): 166-72.
8. Geisert Jr EE, Williams RW, Geisert GR, Fan L, Asbury AM, Maecker HT. Increased brain size and glial cell number in CD81- null mice. J Comp Neurol 2002; 453: 22-32.
9. Kölker S, Christensen E, Leonard JV, et al. Diagnosis and management of glutaric aciduria type I: revised recommendations. J Inherit Metab Dis 2011; 34: 677-94.
10. Seijo-Martínez M, Navarro C, Castro del Río M, et al. L-2-hydroxyglutaric aciduria: clinical, neuroimaging, and neuropathological findings. Arch Neurol 2005; 62: 666-70.
11. van der Knaap MS, Naidu S, Breiter SN, et al. Alexander disease: diagnosis with MR imaging. Am J Neuroradiol 2001; 22: 541-52.
12. Le Coq J, An HJ, Lebrilla C, Viola RE. Characterization of human aspartoacylase: The brain enzyme responsible for Canavan disease. Biochemistry 2006 45: 5878-84.
13. van der Knaap MS, Scheper GC. Megalencephalic leukoencephalopathy with subcortical cysts. En: Pagon RA, Adam MP, Ardinger HH, et al (eds). GeneReviews®. Seattle, WA: University of Washington; 1993-2015. August 11, 2003.
14. Pronk JC, van Kollenburg B, Scheper GC, et al. Vanishing white matter disease: a review with focus on its genetics. Ment Retard Dev Disabil Res Rev 2006; 12: 123-8.
15. Pavone P, Praticò AD, Rizzo R, et al. A clinical review on megalencephaly: A large brain as a possible sign of cerebral impairment. Medicine 2017; 96: e6814.
16. Ruggieri V, Arberas C. Síndromes genéticos reconocibles en el periodo neonatal. Medicina (B Aires) 2009; 1:15-35.
17. Pilarski R, Stephens JA, Noss R, et al. Predicting PTEN mutations: an evaluation of Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome clinical features. J Med Genet 2011; 48: 505-12.
18. Tenorio J, Arias P, Martínez-Glez V, et al. Simpson-Golabi-Behmel syndrome types I and II. Orphanet J Rare Dis 2014; 9: 138.
19. Parker WE, Orlova KA, Parker WH, et al. Rapamycin prevents seizures after depletion of STRADA in a rare neurodevelopmental disorder. Sci Transl Med 2013; 5: 182ra53.
20. Puffenberger EG, Strauss KA, Ramsey KE, et al. Polyhydramnios, megalencephaly and symptomatic epilepsy caused by a homozygous 7-kilobase deletion in LYK5. Brain 2007; 130: 1929-41.
21. Butler MG, Dasouki MJ, Zhou XP, et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet 2005; 42: 318-21.
22. Mirzaa GM, Poduri A. Megalencephaly and hemimegalencephaly: breakthroughs in molecular etiology. Am J Med Genet C Semin Med Genet 2014; 166C: 156-72.
23. Graham JM, Jr, Clark RD, Moeschler JB, et al. Behavioral features in young adults with FG syndrome (Opitz-Kaveggia syndrome). Am J Med Genet C Semin Med Genet 2010; 154C: 477-85.
24. Opitz JM, Kaveggia EG. Studies of malformation syndromes of man 33: the FG syndrome. An X-linked recessive syndrome of multiple congenital anomalies and mental retardation. Z Kinderheilkd 1974; 117: 1-8.
25. Coffee B, Keith K, Albizua I, et al. Incidence of fragile X syndrome by newborn screening for methylated FMR1 DNA. Am J Hum Genet 2009; 85: 503-14.
26. Bagni C, Tassone F, Neri G, Hagerman R. Fragile X syndrome: causes, diagnosis, mechanisms, and therapeutics. J Clin Invest 2012; 122: 4314-22.
27. Szudek J, Birch P, Friedman JM. Growth charts for young children with neurofibromatosis 1 (NF1). Am J Med Genet 2000; 92: 224-8.
28. Costa RM, Silva AJ. Mouse models of neurofibromatosis type I: Bridging the GAP. Trends Mol Med 2003; 9: 19-23.
29. Moore BD III, Slopis JM, Jackson EF, De Winter AE, Leeds NE. Brain volume in children with neurofibromatosis type 1: Relation to neuropsychological status. Neurology 2000; 54: 914-20.
30. Cutting LE, Cooper KL, Koth CW, et al. Megalencephaly in NF1: Predominantly white matter contribution and mitigation by ADHD. Neurology 2002; 59: 1388-94.