
JORGE MALAGON VALDEZ
Universidad Autónoma de Aguascalientes
Resumen El síndrome de West o espasmos infantiles, es una encefalopatía epiléptica clasificada como epi-
lepsias y síndromes generalizados. Hay múltiples informes de la evolución de síndrome de West a síndrome de Lennox-Gastaut de un 25 hasta 60%, sin reconocerse una causa específica. Se ha comunicado que pueden ser solo una entidad epiléptica dependiente de la edad y que estaría en relación con el grado de inmadurez cerebral. En esta revisión retrospectiva de 130 casos de espasmos infantiles, solo 14 (10.7%) evolucionaron a Lennox-Gastaut. El haber recibido en todos los casos vigabatrina como tratamiento nos hace suponer que la baja incidencia podría estar relacionada con el uso de este fármaco. Dado que la vigabatrina tiene una acción gabaérgica y aumenta los niveles de ACTH podría explicar esta relación, pero esto deberá confirmarse con el mejor conocimiento de los mecanismos íntimos de estas graves encefalopatías.
Palabras clave: intersíndrome, síndrome de West, síndrome de Lennox-Gastaut, vigabatrina
Abstract Ttransional syndrome: from West to Lennox-Gastaut syndromes. West syndrome or infantile
spasms is an epileptic encephalopathy, classified as generalized epilepsies and syndromes. There are multiple reports of the evolution from West to Lennox-Gastaut syndrome of 25 up to 60%, without a specific cause is determined. It has been reported that they may be only an epileptic entity age dependent that it would be in relation to the degree of brain immaturity. In this retrospective review of 130 cases of West syndrome, only
14 (10.7%) evolved to Lennox-Gastaut. Having received in all cases vigabatrin as a treatment, makes us suppose that the low incidence could be related to the use of this drug. Given that vigabatrin has a gabaergic action and increased levels of ACTH, may explain this relationship but this must be confirmed with the best knowledge of the intimate mechanisms of these serious epileptic encephalopathies.
Key words: intersyndrome, West syndrome, Lennox-Gastaut syndrome, vigabatrin
e-mail: neuronags@hotmail.com
Los síndromes de West (SW) y Lennox-Gastaut (SLG) se consideran epilepsias catastróficas de inicio temprano. El SW o espasmos infantiles (EI), se inician dentro del primer año de vida con un pico entre los 3 a 9 meses y el SLG entre 1 a 4 años. Es conocida la transición en algunos casos en los que el SW pasa a SLG. Hay teorías que sugieren ser un continuo del mismo proceso patológico porque tienen algunas características clínicas similares1. Las crisis en ambas entidades suelen ser de difícil control, su pronóstico en ambos síndromes es pobre.
Los EI han sido revisados en múltiples ocasiones a través de los años, pero su fisiopatología permanece poco clara. La incidencia no es muy frecuente, se ha determinado en 1 por cada 2000 a 6000 recién nacidos vivos. Sus múltiples etiologías hacen que el tratamiento sea variado y el pronóstico diferente en cada caso. Se caracteriza por una triada con espasmos, retraso psicomotor y un electroencefalograma (EEG) con un trazo denominado hipsarritmia, aunque hay casos en que el EEG no es lo característico de este tipo. Diversas publicaciones los han relacionado. Aproximadamente un 25% a 54% de los casos de EI evolucionan a un SLG2 y los que presentan Lennox-Gastaut tienen antecedente de EI hasta en un 36%. Hay algunas series que no informan ningún caso. El SLG es una encefalopatía epiléptica constituida también por una triada: a) crisis de múltiples tipos como tónicas, tónico-clónicas generalizadas, atónicas, ausencias atípicas y mioclónicas, b) retraso psicomotor y c) EEG característico con descargas de punta onda lenta generalizada de 1.5 a 2.5 Hz, y en sueño descargas de ritmos rápidos3. Es una epilepsia que a menudo suele ser de difícil control. Al igual que en el SW la etiología suele ser variable y su manejo complejo4.
De acuerdo a la última clasificación de la Liga Internacional contra la Epilepsia, estos síndromes se incluyen en el grupo de epilepsias y síndromes generalizados y se dividen en sintomáticos o criptogénicos. El objetivo fue determinar si hay alguna variabilidad clínica, de etiología o tratamiento, que provoque la evolución del SW a SLG.
Materiales y métodos
Se estudiaron retrospectivamente 130 casos de SW en una revisión de historias clínicas por un período de 10 años (2006-2016). Solo 14 (10.7%) del total evolucionaron a Lennox-Gastaut5. Se incluyeron niños con la triada de EI, retraso psicomotor, espasmos infantiles y EEG con hipsarritmia. Once (78.5 %) fueron masculinos. A todos los pacientes se les administró vigabatrina con una duración de 6 meses a un año, previo EEG de control. Solo en el 12.5 % se agregó metilprednisolona, a ninguno se le administró ACTH. Estos pacientes presentaron durante su evolución las características clínicas y EEG de Lennox-Gastaut. Crisis tónicas, tónico-clónicas generalizadas, atónicas, ausencias atípicas y parciales complejas6 y EEG con descargas de punta onda lenta generalizada de menos de 3 Hz, con un retraso psicomotor importante. El cambio de la sintomatología se presentó en promedio a los 2.5 años. La etiología de los pacientes que evolucionaron a Lennox-Gastaut fue considerada sintomática como se muestra en la Tabla 1.
Resultados
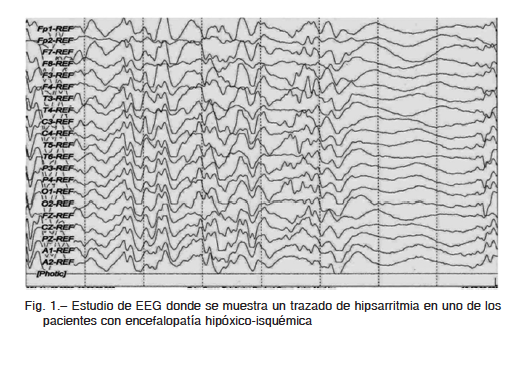
En los 14 pacientes con síndrome de West, las manifestaciones clínicas fueron los espasmos y el promedio de tiempo del diagnóstico fue entre los 9 y 10 meses de edad. Se realizó EEG en el 100% (Fig. 1). Ocho (57%) tenían tomografía computada cerebral y seis (43%) resonancia magnética cerebral. Las crisis predominantes fueron los espasmos en flexión, en algunos casos fueron mixtos. En todos, el tratamiento fue con vigabatrina de 40 a 150 mg/kg/día y ácido valproico asociado 20-40 mg/kg/día7, con excelente respuesta, controlándose los espasmos en el 87% de los casos. Los potenciales evocados visuales fueron normales en todos los pacientes8.
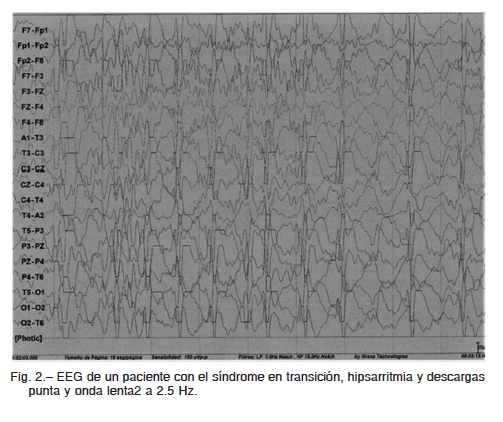
Las crisis en el SLG fueron tónicas en 60%, ausencias atípicas 38%, atónicas 42%, tónico- clónicas generalizadas 31%, presentándose en forma combinada. El diagnóstico se confirmó con EEG en todos (Fig. 2). El tratamiento del SLG fue con múltiples antiepilépticos como ácido valproico, lamotrigina, topiramato, clobazam, metilprednisolona, oxcarbazepina y levetiracetam9. No se utilizó dieta cetógena. No hubo correlación entre el diagnóstico etiológico y el tiempo de las manifestaciones del SW con la evolución a SLG. Por lo tanto, no hubo cambios notables a los descritos en otras series en relación a la clínica, tipos de crisis, etiología, solo la observación del tratamiento con vigabatrina.
Discusión
El porcentaje de casos de espasmos infantiles que evolucionan a síndrome de Lennox Gastaut es variable, del 30 al 60% en diversas series. En nuestra experiencia es menor a lo comunicado en otros estudios. La diferencia más importante que hemos detectado fue el uso de la vigabatrina en todos ellos, solo se utilizaron esteroides en tres casos en forma asociada. No hay informes que indiquen que la vigabatrina disminuya la probabilidad de la evolución a SLG. Se ha sugerido que el uso de
dieta cetógena podría disminuir esta evolución10. Es de destacar que en Colombia, país donde tampoco se cuenta con ACTH, en una revisión de 36 pacientes con SW tratados con ácido valproico y vigabatrina, solo uno evolucionó a SLG11.
El mecanismo de acción de la vigabatrina es gabaérgica, un derivado vinil del g-ácido aminobutírico que se une irreversiblemente en la sinapsis a la gabatransaminasa, enzima que metaboliza el GABA, lo que provoca que se incremente el GABA en el cerebro y de ahí su acción antiepiléptica. La actividad enzimática no se recobra hasta que no se sintetiza una nueva enzima, lo que tarda entre cuatro y seis días12. Se ha comunicado que reduce al tetrapéptido de colecistoquinina, con disminución de los síntomas inducidos por trastorno del pánico, además de elevar los niveles de ACTH y cortisol13. Quizá actúe por este medio también en contra de la inmunoinflamación.
Reciente estudios de epileptogénesis en los EI, incluyen varios procesos como la neuroinflamación, alteraciones del estado inmune y del sistema endócrino. Se ha sugerido a la neuroinmunomodulación como la llave de los mecanismos comprometidos en la epileptogénesis del síndrome de West. El tratamiento hormonal y la dieta cetógena sugiere que juegan un papel importante en la prevención de la evolución de EI a SLG. Sin embargo, no hay pruebas suficientes para demostrar el impacto de estos mecanismos en la evolución de EI a SLG14.
Se ha publicado que los mecanismos inmunológicos tienen un papel preponderante en la transición de SW a SLG, ya que se ha encontrado una deficiencia inmunológica de tipo celular, donde hay inhibición de la migración leucocitaria, además de la trasformación blástica de los linfocitos, así como disminución de linfocitos B y T en sangre periférica y niveles séricos de inmunoglobulinas15.
El uso de la vigabatrina está limitado por sus efectos adversos, sobre todo la constricción permanente del campo visual. Pero por otro lado, el riesgo catastrófico del pronóstico cognitivo y conductual causado por los espasmos sin control, es más grave. Se la considerada el tratamiento de primera línea para los EI, sobre todo en los que la enfermedad de base es la esclerosis tuberosa. Es necesario conocer los diversos efectos secundarios de la vigabatrina como el daño a la retina y las alteraciones al parecer reversibles que se presentan en los ganglios basales, manifestados como hiperintensidad reversible en la resonancia magnética cerebral16. En animales de experimentación, después de un año de administración de dosis altas, se ha descrito un edema intramielínico (microvacuolación) a nivel cerebral (en sustancia blanca) que desaparece a las pocas semanas de retirar el medicamento.
Aún hay debate sobre si EI y SLG es una misma encefalopatía y que las diferentes expresiones dependen del nivel de madurez cerebral. Los supuestos mecanismos antiepilépticos como la inhibición de la inflamación y modificación del metabolismo mitocondrial, así como la acción de la terapia hormonal o dieta cetógena, han sido propuestos como mecanismos que juegan un papel importante en prevenir que los espasmos evolucionen a SLG; sin embargo, no hay pruebas suficientes para confirmar esta aseveración17. La observación de que la vigabatrina puede disminuir el riesgo del paso de EI a SLG es una probabilidad que podría llamar la atención y ser estudiada con mayor profundidad.
Es necesario determinar la fisiopatología de las dos entidades para demostrar si hay o no una relación en su transición, ya que siendo una encefalopatía epiléptica provoca más deterioro neurológico y esto podría darnos como resultado un tratamiento del SW más específico y así poder evitar la evolución a SLG.
Conflicto de intereses: Ninguno para declarar
Bibliografía
1. Ranttala H, Putkonen T. Ocurrence. Outcome and prognostic factors of infantile spasm and Lennox-Gastaut syndrome. Epilepsia 1999; 40: 286-9.
2. Olmos-García de Alba G, Malagon-Valdez J, Valarezo-Crespo F. West syndrome evolving into the Lennox-Gastaut syndrome. Clini Electroencephalogr 1984; 15: 61-8.
3. Karvelas G, Lortie A, Scantlebury MH, Duy PT, Cosette P, Carmant L. A retrospective study on aetiology based outcome of infantile spasm. Seizure 2009; 18: 197-201.
4. Gastaut H, Roger J, Soulayrol R, et al. Epileptic encephalopathy of children with diffuse slow spikes and waves (alias ‘‘petit mal variant’’) or Lennox syndrome. Ann Pediatr 1966; 13: 489-99.
5. Shields WD. Diagnosis of infantile spasms, Lennox-Gastaut syndrome, and progressive myoclonic epilepsy. Epilepsia 2004; 45(Suppl. 5): 2–4.
6. Archila R, Papazian O. Lennox Gastaut Syndrome. Rev Neurol 1999; 29: 346-9.
7. Caraballo RH, Cersósimo R, Arroyo HA, Fejerman N. Síndrome de West sintomático: asociaciones etiológicas particulares con respuesta inesperada al tratamiento. Rev Neurol 1998; 26: 372-5.
8. JonesK, Go C, McCoy B, Puka K, Snead OC 3rd. Vigabatrin as first-line treatment for infantile spasm not related to tuberous sclerosis complex. Pediatr Neurol 2015; 53: 141-5.
9. Díaz-Negrillo, Martín del Valle F, González-Salaices M, Prieto CJ, Carneado Ruiz J. Eficacia del levetiracetam en pacientes con síndrome de Lennox-Gastaut. Presentación de un caso. Neurología 2011; 26: 285-90.
10. You SJ, Kim HD, Kang HC. Factors influencing the evolution of West syndrome to Lennox-Gastaut syndrome. Pediatr Neurol 2009; 41: 111-3.
11. Quintero CA, Sierra G, Fajardo A, Salvatierra I, Espinosa E. Síndrome de West en el Hospital Militar Central y en el Instituto de Ortopedia Infantil Roossevelt: análisis retrospectivo de los casos presentados entre los años de 2002 a 2004. Acta Neurol Colomb 2005; 21: 115-20.
12. Vigabatrina. En: http://salud.es/medicamento/vigabatrina/. 23/06/2009; consultado junio 2018.
13. La vigabatrina, indicaciones, mecanismo de acción, los efectos adversos, interacción con otros medicamentos, farmacocinética. En: etasto.com/caja-de-cerebro/conocimiento-7584.htm; consultado junio 2018.
14. Oleksii S, Solomon L. M, Aristea SG. Inflammation in epileptic encephalopathies. Adv Protein Chem Struct Biol 2017; 108: 59-84.
15. Montelly TC, Iwasso MT, Peracoli MT, Moya NG. Cell-mediated and humoral immunity in West syndrome. Arq Neuropsiquiatr 1981; 39: 1-12.
16. Fernández-García A, García-Peñas JJ, Gómez-Martín H, Pérez-Sebastián I, García-Esparza E, Sirvent-Cerdá S. Alteraciones reversibles en la neuroimagen asociadas al tratamiento con vigabatrina en lactantes con espasmos epilépticos. Rev Neurol 2017; 64:169-74.
17. Shandra O. Does activation of brain inflammatory signaling pathways contribute to the transition of WS to LGS? En: Donev R. Protein Chemistry and Structural Biology. Stress and Inflammation in Disorders. Cambridge, MA, USA: Elsevier 2017.