
JAUME CAMPISTOL
Servicio Neuropediatría, Hospital Universitari Sant Joan de Déu, Barcelona, España
Resumen Los trastornos paroxísticos no epilépticos (TPNE) se definen como episodios que se repiten pe-
riódicamente y que remedan una crisis epiléptica. Su aparición es generalmente brusca y de breve duración, originados por una disfunción cerebral de origen diverso y que no obedecen a una descarga neuronal excesiva (a diferencia de una crisis epiléptica). Su incidencia es diez veces más elevada que la epilepsia y pueden aparecer a cualquier edad, pero son más frecuentes en los primeros años de vida. La inmadurez del sistema nervioso en la infancia hace que en este período las manifestaciones sean muy diversas y diferentes a otras edades. El primer paso para un diagnóstico correcto es un buen interrogatorio y establecer si el episodio puede corresponder a una crisis epiléptica o a un TPNE. El diagnóstico diferencial es muy amplio, especialmente en las primeras edades. Aparte del examen neurológico completo, en caso de duda se debe ampliar el estudio con exámenes complementarios que en la mayoría de las ocasiones serán normales/ negativos. En algunos casos se ha demostrado una base genética. Las opciones terapéuticas son escasas y la mayoría de los TPNE, especialmente en el lactante, desaparecen con la edad sin dejar secuelas.
Palabras clave: trastornos paroxísticos no epilépticos, lactante, protocolo diagnóstico, genética, tratamiento
Abstract Non-epileptic paroxysmal disorders in infant. Non-epileptic paroxysmal disorders (PNED) are
defined as events that mimic epileptic seizures. Its onset is usually sudden and short-lived, caused by brain dysfunction of various origins, but not due to excessive neuronal firing. Its incidence is higher than the epilepsy (10:1). They can occur at any age but are most common in children, especially in the first year of life. The immature nervous system in childhood causes in this period, paroxysmal manifestations that are very diverse and different from other ages. Normal and common paroxysmal disorders in children can mimic epileptic seizures. The first step is to establish a correct diagnosis, if the clinical paroxysmal episode is a seizure or PNED. Differential diagnosis is very broad, especially in the first ages. It’s necessary a complete neurological examination in case of doubt and the study should be extended with complementary exams, investigations that in most cases will be normal/negative. In some of them, a genetic basis has been shown. Treatment options are limited and most PNED untreated have a favorable outcome.
Key words: paroxysmal non epileptic events, infant, diagnosis, genetics, treatment
e-mail: campistol@sjdhospitalbarcelona.org
Los trastornos paroxísticos no epilépticos (TPNE) en el lactante constituyen un grupo de situaciones muy polimorfas desde el punto de vista semiológico, en las que se producen cuadros clínicos muy diversos que pueden remedar una crisis epiléptica y que están causados por procesos fisiológicos, por un desencadenante o, en ocasiones, de origen desconocido. La diferencia con una crisis epiléptica (debida a una descarga neuronal) a veces es muy sutil, en ocasiones difícil de evidenciar y con facilidad se pueden confundir ambos trastornos1. La incidencia de los TPNE en la infancia es mucho más elevada que la epilepsia (10:1). Muchos de estos trastornos, especialmente en el lactante, son edad-dependiente y desaparecen sin dejar secuelas en la mayoría de los casos. Los exámenes complementarios deben ser solicitados e interpretados adecuadamente, siempre teniendo presente al paciente, sus circunstancias y especialmente el cuadro clínico. En general, solamente se solicitan en caso de duda y para descartar otra enfermedad responsable, como puede ser la epilepsia, una enfermedad metabólica, un proceso expansivo o una enfermedad extraneurológica de origen cardíaco o digestivo.
Se debe enfatizar el valor que tiene el trazado EEG frente a un paciente con epilepsia y el limitado valor confirmativo que tiene en un TPNE2. Todo ello nos lleva a concluir que el registro EEG, aun a pesar del gran valor que tiene en el estudio de los cuadros paroxísticos, puede ser una de las principales causas de error (pacientes con epilepsia pueden tener el EEG normal y sin epilepsia mostrar alteraciones en el EEG). No obstante, realizado en buenas condiciones e interpretado adecuadamente, permitirá confirmar el diagnóstico2,3.
Otra causa de error es el desconocimiento que existe de los TPNE del lactante y la facilidad de considerar epiléptico cualquier episodio paroxístico1,3,4. Es importante, aparte de tener en cuenta estas causas de error, disponer de un protocolo de estudio.
Pauta de estudio frente a un lactante con un TPNE
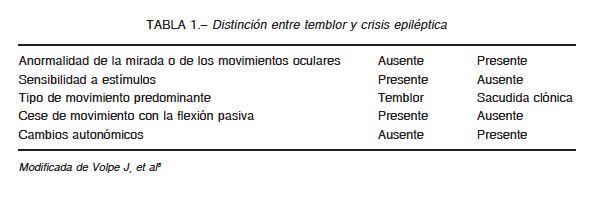
El enfoque frente a un episodio paroxístico en un lactante pasa por un interrogatorio completo con una adecuada valoración de los antecedentes, la exploración física y los exámenes complementarios dirigidos en función del fenómeno presentado. Es de gran utilidad el análisis detallado del episodio en vídeo que casi siempre aportan los padres para su posterior análisis. Determinar el origen epiléptico no siempre es sencillo y puede requerir, en caso de duda una prudente espera, en ocasiones, de un registro video-EEG o de otras exploraciones complementarias muy puntuales (Tabla 1)1.
TPNE en el lactante
Existen muchas clasificaciones de los trastornos paroxísticos no epilépticos en el lactante. Clásicamente se dividen en: TPNE relacionados con el movimiento, la hipoxia, el dolor, el sueño y un grupo de miscelánea. No podemos revisar en esta capítulo todos y cada uno de los 25 TPNE descritos en el lactante (Tabla 2). Existen algunas

monografías al respecto donde se detallan todos ellos1. Nos limitaremos a revisar algunos de los más comunes TPNE del lactante en la práctica diaria, analizando sus características principales y diferenciales, fisiopatogenia, herencia y opciones terapéuticas.
TPNE relacionados con el movimiento
Espasmus nutans: Se caracteriza por episodios espontáneos de nistagmo asimétrico asociado a inclinación y movimientos de afirmación o negación de la cabeza. La duración es de escasos segundos pero tiene tendencia a repetir. El proceso es autolimitado y desaparece entre los 3 y 6 años. El EEG es normal. Hay que descartar la posibilidad remota de lesiones quiasmáticas o diencefálicas1,5.
Estremecimientos: Son eventos paroxísticos breves y frecuentes que se inician hacia los 4 meses pero se extienden a lo largo de la infancia. Consisten en cesación de la actividad sin pérdida de conciencia, temblor brusco rápido de corta duración, especialmente de la cabeza con posturas tónicas con flexión o extensión de la cabeza y cuello con estremecimiento cefálico y de tronco. Puede haber un factor precipitante, repetir varias veces al día y no hay postcrítico. El EEG ictal es normal6.
Desviación paroxística de la mirada hacia arriba con ataxia: El cuadro se inicia entre los 3 meses y 2 años y consiste en episodios prolongados (segundos, horas o días) de desviación continua o episódica de los ojos hacia arriba con ataxia asociada. Durante el episodio, el intento del niño de mirar hacia abajo le induce un nistagmo en la misma dirección y la flexión de la cabeza sobre el tronco para compensar la desviación. Los síntomas desaparecen con el sueño y se incrementan con el cansancio o infecciones. El cuadro remite espontáneamente en unos años y la mitad de los casos evolucionan con retraso del desarrollo y el lenguaje. Se desconoce la causa, algunos casos mejoran con L-dopa. La neuroimagen es normal y existen cuadros familiares7,8.
Reacciones extrapiramidales a fármacos: Pueden aparecer a cualquier edad pero son más comunes en el lactante. Diversos fármacos pueden producir una reacción idiosincrásica aguda y transitoria que en ocasiones asemejan una crisis epiléptica. La semiología más frecuente es la distonía de cuello, musculatura facial y buco-lingual. Los fármacos más implicados son la metoclopramida y los neurolépticos. Responden bien al biperideno. Es importante la historia clínica minuciosa y las determinaciones para identificar y retirar el producto ingerido1.
Vértigo paroxístico benigno: Se presenta entre los 2 y 4 años en forma de episodios breves de inestabilidad sin aura y que se resuelve de forma espontánea en poco tiempo. Puede ser confundido con epilepsia debido a su carácter paroxístico, recurrente, asociado a sensación de miedo y manifestaciones autonómicas (sudoración, náuseas, vómitos). No existe pérdida de conciencia y no hay período postcrítico. La frecuencia de los episodios es muy variable y el cuadro remite hacia los 5 años. Se considera que puede asociarse a migraña1,9,10.
Síndrome de Sandifer: El lactante presenta periódicamente posturas anómalas del cuello, tronco y extremidades como consecuencia de un reflujo gastroesofágico, hernia hiatal o disfunción esofágica. En otras ocasiones puede presentarse en forma de episodios súbitos de tortícolis, rigidez generalizada y opistótonos que pueden acompañarse de apnea, mirada fija con mioclonías de miembros. Suelen ocurrir pocos minutos después de comer y puede no manifestar regurgitaciones, otra característica es la normalidad intercrítica. Se presenta especialmente entre los 18-36 meses. El cuadro puede remedar una distonía o una crisis epiléptica. La presencia de regurgitaciones junto con un EEG crítico normal descarta epilepsia. El cuadro cede al tratar el reflujo gastroesofágico1.
Mioclonías benignas del lactante: Suelen debutar entre los 3 y 9 meses de vida aunque también en el período neonatal. El lactante presenta al despertar salvas repetidas de bruscas contracciones mioclónicas en flexión de cuello y extensión y abducción de los miembros superiores que recuerdan a los espasmos epilépticos del síndrome de West. Se presentan en vigilia y varias veces al día. Pueden ser también sutiles, similares a una mioclonía generalizada. El EEG es normal y no existe deterioro cognitivo. Ceden espontáneamente hacia los 9 meses. Aunque se han descrito con más frecuencia en niños sobrestimulados, se desconoce el origen del trastorno 2, 3, 11, 12.
Crisis tónicas reflejas del lactante: Se presenta en los primeros meses de vida (2-3 m) y se manifiesta por la contracción tónica con extensión de las 4 extremidades, apnea y cianosis, y notable congestión facial durante 3-10 segundos. Se desencadena por estímulos táctiles o cambios posicionales, cuando el niño está en vigilia o desplazado en posición vertical. Es más frecuente en lactantes irritables y en ocasiones aparece en salvas durante 2-3 días. Cede espontáneamente pocos meses después del inicio. El EEG ictal e interictal es normal. Se desconoce su origen1, 13.
Estereotipias: Se trata de conductas motoras repetitivas (orofaciales, cabeza o miembros superiores con movimientos de aleteo) que se presentan de forma rítmica y continuada, en niños por lo demás normales, se acentúan en situaciones de excitación o estrés. Suelen comenzar al año y remiten entre los 2-3 años. Son más acentuados y persistentes en pacientes con enfermedad neurológica (discapacidad intelectual o autismo)1, 9.
Body rocking: Se inicia en el primer año con movimientos de balanceo cuando el niño está en sedestación con movimientos rítmicos de tronco y cabeza tanto en dirección lateral como póstero-anterior. Aunque puede presentarse en niños normales es más común en lactantes poco estimulados, con discapacidad o con déficit sensorial3,4.
TPNE relacionados con la hipoxia
Espasmos del sollozo: Constituye uno de los TPNE más frecuentes en el lactante. Los episodios suelen aparecer entre los 6-18 meses y pueden persistir más tiempo o iniciarse antes. Existen dos tipos: pálido y cianosante. El primero es básicamente un síncope vasovagal por un mecanismo cardioinhibitorio neurológicamente mediado. Se desencadena por un leve e inesperado golpe, especialmente en la cabeza. El niño pierde la conciencia, cae al suelo, se queda pálido e hipotónico. El episodio puede hacer pensar en una crisis si no se detecta que fue precedido por traumatismo y si además se sigue de mioclonías. La recuperación tiene lugar en pocos según dos-minutos. Los episodios remiten con la edad aunque pueden manifestar posteriormente síncopes vasovagales. Algunos niños presentan anemia ferropénica asociada y de dudosa relación con el trastorno. El espasmo del sollozo cianosante tiene su origen en una apnea espiratoria prolongada. El episodio se inicia por llanto o grito seguido de apnea, pérdida de conciencia, cianosis y en ocasiones posturas tónicas. Suele ser desencadenado también por pequeños traumatismos, por miedo o frustración. Son mucho más frecuentes que los pálidos y en ambos tipos se encuentran antecedentes familiares. La evolución es favorable, no precisan tratamiento aparte de identificar el trastorno y tranquilizar a la familia. Existen formas mixtas de espasmos del sollozo3, 14.
TPNE relacionados con el dolor
Vómitos cíclicos del lactante: Se caracterizan por episodios agudos y repetidos de náuseas y vómitos, a menudo asociados a cefalea o dolor abdominal, y que pueden durar varios días. Muchos casos requieren rehidratación parenteral. Estos episodios repiten con cierta periodicidad (3-4 por año) y no se encuentra factor desencadenante. Algunas enfermedades metabólicas de presentación intermitente se pueden manifestar de forma similar. Se inician en el primer año pero pueden persistir varios años. Suelen existir antecedentes familiares de migraña y los mismos pacientes pueden manifestar migraña en edades posteriores1,4.
Dolor paroxístico extremo: Puede debutar ya en período neonatal o especialmente en el lactante con un cuadro de dolor paroxístico intenso especialmente a nivel rectal y en región perianal. Posteriormente, el dolor puede persistir en esta zona o localizarse a nivel ocular, submandibular o submaxilar1,15. Otro hallazgo constante es la rubicundez facial que acompaña al dolor. El fenómeno autonómico se acompaña con frecuencia de rigidez y espasmo tónico; puede manifestar bradicardia y asistolia, conduciendo finalmente a un cuadro sincopal15. Entre las crisis, los pacientes están asintomáticos. Se trata de una canalopatía hereditaria autosómica dominante, debida a una mutación en el gen SCN9A responsable de una canalopatía con ganancia de función16. Pueden responder a la carbamazepina, perdura toda la vida, pero los ataques son más frecuentes e intensos en el periodo de lactante15.
TPNE en relación con el sueño
Episodios de apnea: Se definen por la interrupción de la respiración en sueño durante más de 12-15 segundos. Son frecuentes en los primeros dos meses de vida en niños prematuros y pueden ser secundarios a inmadurez, reflujo gastroesofágico o medicación. Se han observado como primera manifestación de una epilepsia focal aunque la mayoría de los casos son de origen desconocido. Su posible relación con la muerte súbita hace aconsejable una monitorización continua durante los meses posteriores al evento y que se considere como un episodio aparentemente letal1, 4, 14.
Head banging: El episodio consiste en movimientos rítmicos de la cabeza cuando el lactante va a dormirse. La persistencia de estos movimientos durante el sueño se denomina jactatio capitis nocturno. Remiten espontáneamente a los 2-3 años y se observan más frecuentemente y con más intensidad en niños con discapacidad neurológica o sensorial.
Miscelánea
Tortícolis paroxístico: Se inicia en el primer año de vida y remite espontáneamente antes de los 3-5 años. El niño presenta de forma recurrente y subaguda, a veces precedido de movimientos oculares anormales, una postura de tortícolis no doloroso, a menudo desencadenado por cambios en la postura. Durante los episodios el paciente puede aquejar malestar, irritabilidad, agitación, palidez, vómitos o incluso ataxia. Los episodios, que suelen cambiar de un lado a otro en cada evento, pueden durar minutos, horas y a veces días, cediendo espontáneamente y repetir con una frecuencia variable a veces constante. Entre los episodios, la exploración neurológica, neuroimagen y EEG son normales. Es más frecuente en niñas (3:1) y tiene un predominio matutino. Se desconoce la etiología aunque se sospecha una canalopatía (CACNA1A y PRRT2) y en relación con la migraña, ya que en ocasiones el cuadro puede evolucionar hacia vértigo paroxístico, ataxia episódica y cefalea migrañosa1, 3, 5.
Episodios de autoestimulación: Se inician entre los 3 meses y 5 años. Se pueden expresar de formas diferentes: posturas distónicas, gruñidos, diaforesis, agitación, cianosis o palidez, mirada perdida o contorsión pélvica. El paciente puede estimular sus genitales directamente o a través de maniobras de frotamiento de los muslos. Ciertas situaciones favorecen los episodios (aburrimiento, viaje en asiento de coche, cansancio, conciliar el sueño). El episodio cesa al distraer al niño, aunque suele mostrarse contrariado con tendencia a reiniciar la actividad. Pueden confundirse con facilidad con crisis focales complejas. Un video doméstico puede ayudar al diagnóstico. El pronóstico es favorable17.
Hemiplejía alternante de la infancia: Este raro fenómeno debuta antes de los 18 meses. Consiste en episodios de hemiplejía flácida subaguda asociados a síntomas autonómicos y nistagmo monocular ipsilateral a la hemiplejía. El episodio se suele iniciar con posturas tónicas o distónicas de miembros con agitación y sensación de miedo. Se sigue de desviación de la cabeza hacia el hemicuerpo afectado y progresa hacia una hemiplejía completa con dificultad para la deglución y respiración. La hemiplejía puede durar desde pocos minutos a varias horas, puede afectar ambos lados de forma simultánea y remite espontáneamente para volver a repetir en el mismo o en el otro hemicuerpo al cabo de pocas semanas. El paciente muestra recuperación completa tras el sueño. Los episodios iniciales pueden ser confundidos con crisis parciales complejas con paresia de Todd o con episodios isquémicos transitorios. El EEG crítico muestra ondas lentas unilaterales en el hemisferio afecto. La evolución es tórpida y los episodios tienden a remitir con la edad; pero los pacientes evolucionan hacia un retraso cognitivo, ataxia, epilepsia, coreoatetosis y problemas cardíacos. Se ha identificado un gen responsable, ATP1A3. Mejoran parcialmente con flunarizina, pero no evita los ataques ni el deterioro neurológico18.
Conflicto de intereses: Ninguno para declarar
Bibliografía
1. Campistol Plana J. Trastornos paroxísticos no epilépticos en la infancia. Barcelona: Viguera Editores, 2014.
2. Fejerman N, Medina CS, Caraballo RN. Trastornos paroxísticos y síntomas episódicos no epilépticos. En: Fejerman N, Fernández Álvarez E, eds. Neurología Pediátrica, 3a ed. Buenos Aires: Panamericana, 2007, p 670-4.
3. Panayiotopoulos CP. Imitators of epileptic seizures. En: Panayiotopoulos CP. A Clinical Guide to Epileptic Syndromes and their Treatment, 2nd ed. London: Springer-Verlag, 2007, p 79-117.
4. Obeid M, Mikati MA. Expanding spectrum of paroxysmal events in children: potential mimickers of epilepsy. Pediatr Neurol 2007; 37:309-16.
5. Vigevano F. Non-epileptic paroxysmal disorders in the first year of life. En: Panayiotopoulos CP , ed. A practical guide to childhood epilepsies. Oxford: Medicinae, 2006, p 107-14.
6. Riehl JA, Mink JW. Shuddering attacks. J Pediatr Neurol 2010; 8: 25-9.
7. Campistol J, Prats JM, Garaizar C. Benign paroxysmal tonic upgaze of childhood with ataxia. A neuro-ophthalmological syndrome of familial origin? Dev Med Child Neurol 1993; 35: 4
8. Zaferiou DI. Paroxysmal tonic upward gaze of childhood “plus”: an oculomotor chanelopathy? Eur J Pediatr Neurol 2015; 19: 278-9.
9. Fernández-Alvarez E. Transient benign paroxysmal movement disorders in infancy. Eur J Pediatr Neurol 2018; 22: 230-7.
10. Kutluay E, Selwa L, Minecan D, Edwards J, Beydoun A. Nonepileptic paroxysmal events in a pediatric population. Epilepsy Behav 2010; 17: 272-5.
11. Fejerman N. Nonepileptic disorders imitating generalized idiopathic epilepsies. Epilepsia 2005; 46 Suppl 9: 80-3.
12. Caraballo RH, Capovilla G, Vigevano F, Beccaria F, Specchio N, Fejerman N. The spectrum of benign myoclonus of early infancy: Clinical and neurophysiologic features in 102 patients. Epilepsia 2009; 50: 1176-83.
13. Vigevano F, Lispi ML. Tonic reflex seizures of early infancy: an age-related non-epileptic paroxysmal disorder. Epil Disord 2001, 3: 133-6.
14. Stephenson JB. Anoxic seizures: self-terminating syncopes. Epileptic Disord 2001; 3: 3-6.
15. Fertleman CR, Ferrie CD, Aicardi J, et al. Paroxysmal extreme pain disorder (previously familial rectal pain syndrome). Neurology 2007; 69: 586-95.
16. Fischer TZ, Waxman SG. Familial pain syndromes from mutations of the NaV1.7 sodium channel. Ann N Y Acad Sci 2010; 1184: 196-207.
17. Rödöö P, Hellberg D. Girls who masturbate in early infancy: diagnostics, natural course and a long-term follow-up. Acta Paediatr 2013; 102: 762-6.
18. Panagiotakaki E, Gobbi G, Neville B, et al. Evidence of a non-progressive course of alternating hemiplegia of childhood: study of a large cohort of children and adults. Brain 2010; 133: 3598-610.