
LUISINA B. PERUZZO1, MARÍA C. BERTONE1, MARINA D. MATHEY1, JORGE G. ROSSI2, EDGARDO M. BAIALARDO3, MARÍA S. FELICE1
1Servicio de Hematología y Oncología, 2Servicio de Inmunología y Reumatología, 3Servicio de Genética, Hospital de Pediatría Prof. Dr. Juan P. Garrahan, Buenos Aires, Argentina
Resumen La leucemia aguda es la enfermedad oncológica más frecuente en la infancia. La leucemia linfoblástica aguda representa el 75% y la mieloblástica aguda el 25% de ellas. La eritroleucemia es una entidad infrecuente, representando menos del 5% de las leucemias mieloblásticas agudas. Su definición ha variado a lo largo del tiempo. La OMS en 2017 define el subtipo de eritroleucemia cuando el porcentaje de eritroblastos representa el 80% de la celularidad total de la médula ósea. El presente trabajo, de tipo analítico, retrospectivo, tuvo como finalidad revisar los hallazgos de morfología, citometría de flujo, citogenética, respuesta al tratamiento y evolución de los casos previamente definidos como eritroleucemia, en nuestro centro, en los últimos 25 años y reclasificar aquellos que no cumplían con los nuevos criterios de la OMS 2017. Entre enero de 1990 y diciembre de 2015, se diagnosticaron 576 casos de leucemia mieloblástica aguda siendo 11 (1.9%) de ellos clasificados como eritroleucemia. Resultaron evaluables 10 casos. La distribución por sexo fue 1:1 y la edad mediana fue 5 (rango: 0.9-14) años. Seis pacientes presentaban antecedentes de síndrome mielodisplásico. Según los nuevos criterios, ninguno de los casos analizados puede ser actualmente definido como eritroleucemia. De acuerdo a la recategorización, fueron definidos como leucemias de subtipos de mal pronóstico, como leucemia aguda indiferenciada, sin diferenciación y megacarioblástica. Solo dos pacientes se encuentran libres de enfermedad, probablemente debido a estos subtipos desfavorables, sumado al antecedente frecuente de mielodisplasia.
Palabras clave: eritroleucemia, criterios diagnósticos, clasificación
Abstract Erythroleukemias. Recategorization according to the World Health Organization´s Criteria.
Acute leukemia is the most frequent malignant disease in childhood. Acute lymphoblastic leukemia represents 75% and acute myeloblastic leukemia 25% of them. Erythroleukemia is a rare entity, corresponding to less than 5% of acute myeloblastic leukemia. Its definition has changed over the time. WHO in 2017 defines erythroleukemia when the percentage of erythroblasts represent 80% of the total cellularity of the bone marrow aspirate. This analytical and retrospective study was performed with the aim of reviewing morphology, flow cytometry and cytogenetic features, response to treatment and outcome of cases previously defined as erythroleukemia in our center during the last 25 years and, in addition to reclassify those cases which do not meet the new WHO 2017 criteria. From January 1990 to December 2015, 576 patients were diagnosed as acute myeloblastic leukemia and 11 (1.9%) of them were classified as erythroleukemia. Ten cases were evaluable. Sex distribution was 1:1 and the median age at diagnosis was 5 (range: 0.9-14) years. Six of them had presented with previous myelodysplastic syndrome. None of the analyzed cases can be currently defined as erythroleukemia, according to the new criteria. When reclassified, the cases were defined as leukemias of subsets with poor prognosis such as acute undifferentiated leukemia, without differentiation and megakaryoblastic leukemia. Only 2 patients remain leukemia-free and this could be explained both by the unfavorable prognosis of these leukemia subtypes, and the antecedent of myelodysplastic syndrome in most of the cases.
Key words: erythroleukemia, diagnostic criteria, classification
Dirección postal: Dra. Luisina B. Peruzzo, Hospital de Pediatría Prof. Dr. Juan P. Garrahan, Combate de los Pozos 1881, 1245 Buenos Aires, Argentina
La leucemia aguda (LA) es la enfermedad oncológica más frecuente en la niñez, representando el 37.3% de las enfermedades malignas en menores de 15 años de acuer do a los datos del Registro Oncopediátrico Hospitalario Argentino1. La leucemia linfoblástica aguda representa el 75% aproximadamente y la leucemia mieloblástica aguda (LMA) alrededor del 25% de las LA2.La definición de LMA engloba a una gran cantidad de neoplasias hematológicas malignas de los precursores mieloides que han sido clasificadas en diferentes sub- grupos. La clasificación del grupo Franco-Americano y Británico (FAB)3 que tomó en cuenta solo la morfología, dividió las LMA en 8 subtipos. La OMS clasificó a los diferentes subtipos de LMA de acuerdo a sus caracte- rísticas citogenéticas, moleculares y de inmunofenotipo, y definió un grupo como LMA no especificadas de otro modo (NOS). Entre ellas se encuentra la eritroleucemia que corresponde al subtipo FAB-M64.
La LMA FAB-M6 es una entidad infrecuente, que representa menos del 5% de las LMA2 y se caracteriza por su evolución tórpida y resistencia al tratamiento, lo que confiere a la enfermedad un pronóstico ominoso.
La definición de leucemia de linaje eritroide ha sido controvertida y ha variado con el correr del tiempo. Inicialmente conocida como “enfermedad de Di Gu- glielmo”5, en el año 2001 la OMS incorporó todos sus aspectos diagnósticos incluyendo la morfología, el inmunofenotipo, las alteraciones citogenéticas y las manifestaciones clínicas, y en 2008 la eritroleucemia se definió como la neoplasia mieloide en la que el porcentaje de precursores eritroides representaba el 50% o más de todas las células nucleadas de la médula ósea con un 20% de mieloblastos6.
En la actualización de la OMS, 2017, se postulan nuevos criterios diagnósticos, con una definición más estricta, estableciendo el diagnóstico de LMA M6 cuando el porcentaje de precursores eritroides representa el 80% de la celularidad total en la médula ósea7.
El presente trabajo tuvo la finalidad de revisar los aspectos morfológicos, de inmunofenotipo, citogenética, la respuesta al tratamiento y evolución de los casos que previamente habían sido definidos como LMA FAB-M6 en el Hospital de Pediatría Prof. Dr. Juan P. Garrahan en los últimos 25 años. Además, otro objetivo fue redefinir los casos que no cumplían con los nuevos criterios de LMA FAB-M6 para la OMS 2017, y reclasificarlos en el subtipo correspondiente.
Materiales y métodos
El presente es un estudio analítico, retrospectivo. La infor- mación fue tomada de la base de datos de LMA del Servicio de Hematología y Oncología, del laboratorio de citometría de flujo y del laboratorio de citogenética del Hospital de Pediatría Juan P. Garrahan.
En el presente estudio se incluyeron a todos los pacientes menores de 16 años con diagnóstico de LMA M6 desde enero
1990 a diciembre 2015, y se excluyeron aquellos casos en los que no se contaba con datos necesarios para la revisión, especialmente aquellos de los que no se disponía de exten- didos de médula ósea.
Se revisaron los extendidos de aspirados de médula ósea fijados y coloreados en May-Grünwald-Giemsa y otras co- loraciones citoquímicas necesarias para la definición de la presencia de mieloblastos. Se realizó el recuento de eritro- blastos y de blastos mieloides en las muestras de 10 casos, disponibles y evaluables. Se aplicaron los criterios de la OMS
2017 para definir eritroleucemia (> 80% de eritroblastos en aspirado de medula ósea). Dicha observación se realizó sobre los aspirados de médula ósea, realizando un conteo de entre 200 y 500 células nucleadas.
Se realizó el análisis de los hallazgos del inmunofenotipo de los casos revisados. En una primera etapa, de 1990 a
1995, dichos estudios se realizaban por microscopía de fluorescencia y a partir de 1995 por citometría de flujo. El panel de estudio fue amplio en la mayoría de los casos, si bien en algunas etapas no se contaba con todos los anticuerpos monoclonales necesarios8.
Se revisaron los hallazgos de estudios citogenéticos de- rivados del método de bandeo G realizados por la técnica previamente descripta9. Cabe destacar que en los casos diagnosticados después del 2002 se contó con estudios de biología molecular (RT-PCR) que permitieron el estudio de alteraciones moleculares de LMA. En la presente revisión en 2 casos se evidenció presencia de trisomía del cromosoma 8 y en un caso se halló deleción del cromosoma 7, ambas alteraciones genéticas relacionadas a mal pronóstico. A su vez, los casos restantes presentaron estudios citogenéticos que pueden ser definidos como complejos, los cuales también se encuentran relacionados a mal pronóstico.
Las variables analizadas fueron: sexo, edad, recuento de leucocitos, nivel de hemoglobina y recuento de plaquetas al momento del diagnóstico de la LMA. Se consideró la mediana y el rango de las mismas. También se investigó el antece- dente de SMD en etapas previas al diagnóstico de LMA y la existencia de características clínicas relevantes o distintivas en esos casos.
Se evaluó la respuesta inicial al tratamiento, considerando la tasa de remisión completa (RC), la de respuesta nula o la muerte durante la fase de inducción.
En los pacientes que alcanzaron la RC se analizaron los eventos adversos que presentaron, considerándose como ta- les la recaída de enfermedad, la muerte en RC, la ocurrencia de una segunda enfermedad maligna y muerte por causa no relacionada a enfermedad oncológica.
Se garantizó la confidencialidad respecto a los datos perso- nales de cada paciente (ley nacional 25.326 de Habeas Data), los cuales fueron registrados en la base datos de manera codi- ficada. Este estudio se rigió por las normas de la Declaración de Helsinki, la resolución nacional nro. 1480/2011 del sistema de evaluación, registro y fiscalización de las investigaciones en salud. Los protocolos de tratamiento administrados fueron aprobados por el Comité de Ética del Hospital de Pediatría Prof. Dr. Juan P. Garrahan.
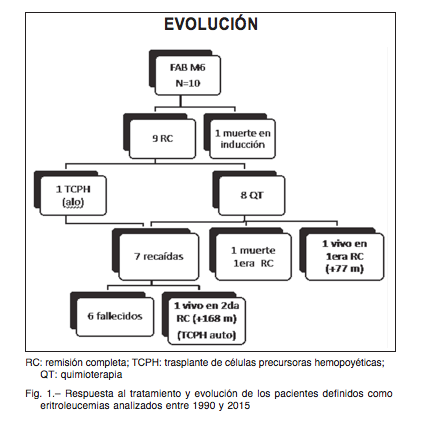
Desde enero de 1990 a diciembre de 2015, ingresaron al Servicio de Hematología y Oncología del Hospital de Pediatría Juan P. Garrahan 576 pacientes con diagnóstico de LMA, de los que 11 fueron categorizados como LMA FAB-M6, lo que representaba el 1.9% de los casos. Todos los pacientes fueron tratados con 4 protocolos sucesivos para LMA adaptados de la estrategia del grupo alemán Berlín-Frankfurt-Müster (BFM)10, 11. De los 11 casos diagnosticados como LMA FAB-M6, 10 cumplieron con los criterios de inclusión. Un caso fue excluido por no disponerse del material de revisión para el diagnóstico morfológico (extendido del aspirado de médula ósea no disponible).
La distribución por sexo de estos 10 pacientes fue 1:1 y la edad mediana al momento del diagnóstico de 5 años (rango: 9 meses-14 años). La mediana del recuento de leucocitos al momento del diagnóstico fue de 6100 (rango:
1200-11 800) /mm3), la mediana del nivel de hemoglobina fue 7.15 (rango: 5.7-10.1) g/dl y la mediana del recuento de plaquetas 59 000 (rango: 25 000-154 000) /mm3.
Al examen físico 4 de los 10 pacientes se presentaron con hepato-esplenomegalia y solo uno presentaba com- promiso extramedular (sarcoma mieloide).
Entre los antecedentes patológicos, seis casos tenían historia de SMD previo al diagnóstico de LMA, detectado en el contexto de diversos grados de citopenia, con gra- dos variables de organomegalia de meses de evolución.
De los 10 casos evaluables, 9 alcanzaron la RC y un paciente falleció durante la fase de inducción, por lo que la tasa de respuesta al tratamiento fue de 90%.
De esos 9 pacientes, solo uno recibió trasplante de células precursoras hemopoyéticas (TCPH) alogéneico en la primera RC. El TCPH fue un trasplante cuyo donante fue un hermano HLA idéntico. Es importante tener pre- sente que el bajo número de trasplantes se debe a que esta es una revisión de 25 años, y que en los primeros años de la misma la oportunidad de ofrecer TCPH con donantes alternativos no era factible en nuestro país. De los 9 casos con RC, 7 recayeron (incluyendo el paciente que recibió TCPH alogéneico en primera RC). El patrón de recaída fue en los 7 casos en médula ósea, mediana en tiempo de recaída: 21 meses desde el diagnóstico (rango: 3-59 meses).
De los 7 casos con recaída solo se rescató uno con un esquema de quimioterapia de segunda línea conso- lidándose luego el tratamiento con un TCPH autólogo. Este enfermo permanece en RC (168 meses desde el diagnóstico). El resto de los pacientes con recaída falle- cieron debido a progresión de enfermedad, por lo tanto solo permanecen vivos y libres de enfermedad dos de ellos: uno en primera RC, que solo recibió tratamiento quimioterápico y otro, que alcanzó una segunda RC y se consolidó con un TCPH autólogo (Fig. 1).
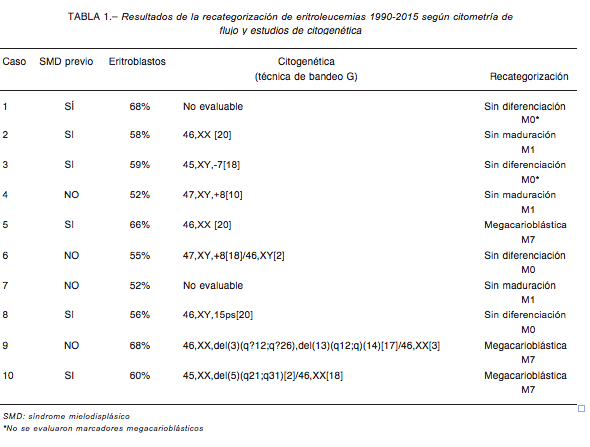
Según la revisión morfológica de los casos previamente diagnosticados como LMA FAB-M6, en ningún caso el porcentaje de eritroblastos alcanzó el 80% requerido de acuerdo a los nuevos criterios de la OMS
2017 para el diagnóstico de eritroleucemia. La mediana de porcentaje de blastos observado fue de 58.5%
(rango: 52-68).
A este análisis morfológico se sumaron los resultados del inmunofenotipo y estudios citogenéticos para lograr recategorizar a estos casos. De dicho análisis surgió que, del total de los casos, tres fueron recategorizados como LMA sin diferenciación (M0), otros tres fueron reclasifi- cados como LMA megacarioblástica (M7), y los cuatro restantes como LMA sin maduración (M1). Cabe destacar, que en dos de los tres casos reclasificados como M0 no se analizaron los marcadores plaquetarios por no contar
con el reactivo para el estudio en la Institución, al momento del diagnóstico de estos casos (Tabla 1).
Discusión
Entre las LMA en pediatría la LMA FAB-M6 representa un subtipo inusual y su definición ha sido controvertida a lo largo del tiempo. En las actualizaciones OMS 2001 y 2008, se consideraron los aspectos clínicos, morfológicos, inmunofenotípicos y genéticos. La definición de la OMS 2017 surgió en el contexto de describir esta enfermedad en relación a su pronóstico y evolución y con criterios más estrictos de clasificación.
Los nuevos criterios de la OMS 2017 definen como LMA FAB-M6 los casos en que el porcentaje de eritro- blastos representa el 80% de la celularidad total en la médula ósea7. La razón de esta modificación se basa en la dificultad para reproducir el conteo de blastos y elementos eritroides, las alteraciones morfológicas transitorias de los precursores de la serie roja y la ausencia de alteraciones genéticas específicas.
Por otro lado, una razón aún más relevante es la gran semejanza clínico-biológica de esta enfermedad con los
SMD12. Tal es así, que en la mayoría de los estudios de reclasificación de casos previamente diagnosticados como LMA FAB-M6, se recategoriza a esta enfermedad como LMA NOS o SMD. De hecho, de los 10 casos es- tudiados, seis presentaban historia de SMD al momento del diagnóstico de LMA.
En los SMD la evidencia del predominio de la serie eritroide podría deberse a una desregulación de diver- sos genes (epigenética), lo que alteraría la expresión de ferritina y la distribución de hierro intramedular, aumen- tando la sensibilidad a la eritropoyetina y produciendo un incremento de la apoptosis y proliferación celular acelerada. Esto conduciría a una diferenciación eritroide ineficaz, lo que indicaría un mecanismo de eritropoyesis inefectiva. Por otro lado, en las denominadas LMA NOS con predominio eritroide, el mecanismo asociado sería la disminución o inefectividad en la granulopoyesis13.
En trabajos recientes se demostró que el mejor factor para predecir la evolución de esta entidad es- taría determinado por las alteraciones citogenéticas y moleculares encontradas en las células neoplásicas, más el porcentaje de blastos en el aspirado de médula ósea, pero hasta ahora no se han descripto alteraciones
citogenético-moleculares recurrentes asociadas a la eritroleucemia.
En un estudio reciente y con el objetivo de la revisión citogenética y de evaluación del pronóstico, Cervera y col. analizaron retrospectivamente 51 casos de leuce- mia clasificada previamente como LMA FAB-M6. Los autores concluyeron que las neoplasias mieloides con predominio eritrocitario muestran un perfil citogenético diferente de aquellas que no tienen predominio de la serie eritroide, dado que se evidenciaron más mutacio- nes en TP53 y menos mutaciones de genes como el DNMT3A y AXSL1. Estos resultados se correlacionan con una peor evolución de los casos de leucemia con predominio de eritroblastos. Así, el pronóstico de esta enfermedad podría correlacionarse de mejor manera con las alteraciones moleculares que con la clasificación fenotípica14.
En el presente estudio las alteraciones citogenéticas detectadas se encuentran relacionadas con subtipos de LMA de pronóstico desfavorable.
Shaowei Qiu y col. analizaron 97 casos de leucemia previamente diagnosticada como LMA FAB-M6 y las reclasificaron según los nuevos criterios de OMS 2017. Los resultados arrojaron que el 67% de los casos se recategorizaron como SMD y el 33% restante como LMA del subgrupo NOS. La supervivencia global de los pacientes con SMD y LMA NOS a los 3 años fue de 56% y 64% respectivamente, lo que no muestra una diferencia significativa. La presencia de anomalías genéticas desfavorables, como la mutación del gen TP53 y RUNX1 se correlacionó con una menor super- vivencia global a los 3 años. Esto demostraría que las alteraciones moleculares definirían mejor el pronóstico de la enfermedad. A su vez, concluyeron que los pa- cientes con factores genéticos desfavorables se verían beneficiados con trasplante de células progenitoras hematopoyéticas (supervivencia libre de enfermedad a los 5 años: 60%)15.
Dentro de la reclasificación de los casos diagnosticados como LMA FAB-M6 en los últimos 25 años en el Hospital de Pediatría Juan P. Garrahan, ninguno de los analizados cumple con los criterios morfológicos necesa- rios para el diagnóstico de esta enfermedad. Los hallazgos en el estudio citogenético no muestran asociación con anomalías citogenéticas recurrentes. Este hallazgo re- fuerza la idea de lo infrecuente de este subtipo de LMA en pediatría, teniendo en cuenta que se trata de una revisión de un largo lapso y en un centro de referencia nacional.
La reclasificación de estos casos según la citometría de flujo los redefine como leucemias de subgrupos de mal pronóstico, como LMA M0, M1 y M7, lo que explicaría los resultados desfavorables en este grupo, junto con el antecedente de cuadro de mielodisplasia previa en el 60% de los casos analizados, que corresponden a un subtipo especial de LMA con pronóstico ominoso.
Conflicto de intereses: Ninguno para declarar
References
1. Moreno F. Registro Oncopediátrico Hospitalario Argentino (5ta. Edición). Incidencia 2000-2013, Supervivencia 2000-2009, Tendencia de Incidencia 2000-2013. En: http://www.msal.gob.ar/images/stories/bes/graficos/0000000730cnt62publicacion-roha.pdf; consultado julio 2017.
2. Pizzo PA, Poplack D. Principles and practice of pediatric oncology. Philadelphia: Lippincott Williams &Wilkins, 2015.
3. Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classification of the acute leukaemias. French-American- British (FAB) co-operative group. Br J Haematol 1976; 33: 451-8.
4. Jaffe ES, Harris NL, Stein H, W. Vardiman (eds). World Health Organization pathology and genetics of tumours of the haematopoietic and lymphoid tissues, 4th ed. Lyon: IARC Press, 2008.
5. Kasyan A, Jeffrey Medeiros L, Zuo Z, et Acute erythroid leukemia as definde in the World Health Organization classification is a rare and pathogenetically heteroge- neous disease. Mod Pathol 2010; 23: 1113-26.
6. Swerdlow SH, Campo E, Harris NL, et WHO classifica- tion of tumours of haematopoietic and lymphoid tissues. Lyon: IARC Press, 2008.
7. Swerdlow SH, Campo E, Lee Harris N, et al. WHO Clas- sification of tumours of haematopoietic and lymphoid tissues. Lyon: International Agency for Research on Cancer,
8. Bene MC, Castoldi G, Knapp W, et al. European Group for the Immunological Characterization of Leukemias (EGIL): Proposal for the immunological classification of acute leukemias. Leukemia 1995; 9: 1783-6
9. Shaffer LG, McGowan-Jordan J, Schmid M. ISCN: An In- ternational System for Human Cytogenetic Nomenclature. Basel (Switzerland): S. Karger AG,
10. Creutzig U, Zimmermann M, Ritter J, et al. Treatment strategies and long-term results in pediatric patients treated in four consecutive AMLBFM trials. Leukemia 2005; 19:2030-42.
11. Felice MS, Rossi JG, Alonso CN, et al. Experience with four consecutive BFM-based protocols for treatment of childhood with non-promyelocytic acute myeloblastic leukemia in Argentina. Leuk Lymphoma 2016; 57: 2090-9.
12. Chen Y, Pourabdollah M, Atenafu EG, et Re-evaluation of acute erythroid leukemia according to the 2016 WHO classification. Leuk Res 2017; 61: 39-43.
13. Zuo Z, Medeiros LJ, Chen Z, et Acute myeloid leukemia (AML) with erythroid predominance exhibits clinical and molecular characteristics that differ from other types of AML. PLoS One 2012; 7: e41485.
14. Cervera N, Carbuccia N, Mozziconacci MJ, et al. Revisiting gene mutations and prognosis of ex-M6 acute erythroid leukemia with regard to the new WHO Blood Cancer J 2017; 7: 1-5.
15. Qiu S, Jiang E, Wei H, et al. An analysis of 97 previously di- agnosed de novo adult acute erythroid leukemia patients following the 2016 revision to World Health Organization classification. BMC Cancer 2017; 17: