
CARLOS ORTEZ, DANIEL NATERA DE BENITO, LAURA CARRERA GARCÍA, JESSICA EXPÓSITO,
GREGORIO NOLASCO, ANDRÉS NASCIMENTO
Unidad de Patología Neuromuscular, Servicio de Neurología Pediátrica, Hospital Sant Joan de Déu, Universidad de Barcelona, España
Resumen La distrofia muscular de Duchenne es una enfermedad genéticamente determinada, ligada al cromosoma X y caracterizada clínicamente por producir debilidad muscular progresiva, con una incidencia de 1 por cada 3500-6000 varones nacidos. Es causada por la mutaciones en el gen DMD, el cual codifica la distrofina, una proteína sub-sarcolémica esencial para la estabilidad estructural del músculo. Los defectos genéticos en el gen DMD, se dividen en: deleciones (65%) duplicaciones (5-10%) y mutaciones puntuales (10–15%). Actualmente no se dispone de tratamiento curativo, el único fármaco que ha demostrado modificar la historia natural de la enfermedad (independientemente de la mutación genética) son los corticoides, los cuales están indicados en estadios tempranos de la enfermedad. En relación a los ensayos clínicos, en los últimos diez años se han experimentado grandes avances en el campo de las opciones terapéuticas, divididos en dos grandes dianas terapéuticas: 1) el área de las terapias génicas y 2) tratar de revertir o bloquear los procesos fisiopatológicos de la enfermedad, tales como inflamación, fibrosis, regeneración muscular, etc. Es probable que un tratamiento eficaz para la distrofia muscular de Duchenne requiera combinaciones que se apliquen tanto al defecto primario como las consecuencias fisiopatológicas secundarias.
Palabras clave: distrofia muscular de Duchenne, avances terapéuticos, terapia génica
Abstract Advances in the treatment of Duchenne muscular dystrophy. Duchenne muscular dystrophy is a genetically determined disease, linked to the X chromosome, c haracterized clinically by producing progressive muscle weakness, with an incidence of 1 per 3500-6000 males born. It is caused by the mutation of the DMD gene, which encodes dystrophin, a sub-sarcolemmal protein essential for structural muscle stability. The genetic defects in the DMD gene are divided into: deletions (65%) duplications (5.10%) and point mutations (10-15%). At present there is no curative treatment, the only drug that has been shown to modify the natural history of the disease (independently of the genetic mutation) are corticosteroids, currently indicated in early stages of the disease. In relation to clinical trials, in the last ten years, has experienced great advances in the field of therapeutic options, divided into two major therapeutic targets: 1) the area of gene therapies and 2) trying to reverse or block the pathophysiological processes of the disease, such as inflammation, fibrosis, muscle regeneration, etc. It is likely that an effective treatment for Duchenne muscular dystrophy requires combinations of therapies that address both the primary defect and its secondary pathophysiological consequences.
Key words: Duchenne muscular dystrophy, therapeutic advances, gene therap
e-mail: ciortez@sjdhospitalbarcelona.org
La distrofia muscular de Duchenne (DMD) es una enfermedad autosómica recesiva ligada al cromosoma X con una incidencia de 1 por cada 3500-6000 varones.
Es causada por mutaciones en el gen DMD que produce la proteína distrofina, una proteína sub-sarcolémica que sirve de anclaje entre el citoesqueleto y la membrana plasmática, por tanto es esencial para la estabilidad estructural del músculo. La distrofina, además de su presencia en el músculo estriado, se expresa en múltiples órganos, entre ellos el cerebro, por lo que pacientes con mutaciones en el gen DMD pueden presentar manifestaciones clínicas como retardo cognitivo, dificultades de aprendizaje, trastornos del espectro autista, etc. Los defectos genéticos en el gen DMD se dividen en: deleciones (65%), duplicaciones (5-10%) y mutaciones puntuales (10-15%) 1-3.
Desde el punto de vista clínico, las mutaciones en el gen DMD producen tres fenotipos: a) DMD cuyos síntomas iniciales aparecen entre los 2 y 3 años de edad, a nivel histopatológico hay usualmente ausencia total de distrofina, b) distrofia muscular de Becker (DMB), en este caso, los síntomas aparecen de manera tardía, a partir de los 5-6 años de edad (incluso hasta la edad adulta), en este subgrupo existe una expresión parcial de distrofina en el musculo, c) distrofinopatías, en este subgrupo se incluyen múltiples manifestaciones clínicas, muchas de ellas no relacionadas con la debilidad muscular, tales como: rabdomiolisis, intolerancia al ejercicio, cardiomiopatía sin debilidad, retardo cognitivo, etc. Al igual que en la DMB, la expresión de distrofina en el musculo está presente de manera parcial.
La historia natural clásica de los pacientes con DMD, se subdivide en cinco períodos clínicos:
a) Pre sintomático muscular o pre debilidad (0-2 años): usualmente no hay manifestaciones clínicas de debilidad, pero suele haber retraso de lenguaje, dificultades de relación-comunicación, retraso motriz global.
b) Ambulante inicial (2-3 años): se producen caídas frecuentes, dificultad para subir escalera sin apoyo, y generalmente, ya se puede apreciar la pseudohipertrofia de gemelos.
c) Ambulante tardío (5-12 años): en este período se manifiestan lo signos clásicos de la DMD: signo de Gowers positivo, pseudohipertrofia muscular, debilidad de cintura pélvica.
d) No ambulante inicial (12-16 años): hay progresión de la debilidad a extremidad superior, inicio de escoliosis, pérdida de la marcha (esta última dependerá de múltiples factores: tipo de mutación, rehabilitación, el uso de corticoides u otras terapias que modifican el tiempo de aparición de pérdida de la marcha, etc.).
e) No ambulante tardío (20 años): fase de silla de ruedas, ventilación pulmonar no invasiva, insuficiencia cardiaca progresiva.
La sospecha diagnóstica de la DMD/B se basa en el cuadro clínico, la historia familiar, los resultados de laboratorio (niveles elevados de creatina-quinasa sérica) y los hallazgos miopáticos en la electromiografía. La biopsia muscular muestra signos de distrofia caracterizados por necrosis y signos de regeneración muscular, además de la ausencia total de distrofina en DMD y ausencia parcial en DMB y distrofinopatías.
El 85-90% del diagnóstico genético de DMD/B se realiza mediante la técnica MLPA (multiplex ligationdependent probe amplification) que detecta deleciones y duplicaciones. Las mutaciones puntuales (15–10%) se detectan mediante secuenciación directa del gen DMD o por secuenciación de exoma 1-4.
Actualmente no se dispone de tratamiento curativo para la DMD/B, el tratamiento integral es sintomático y
sobre todo orientado a la prevención de complicaciones tempranas de la enfermedad (escoliosis, retracciones asimétricas, insuficiencia cardiaca y respiratoria, etc.). El uso de corticoides en DMD ha demostrado que previenen la escoliosis y hay una prolongación de la marcha con el uso estos (en promedio, pacientes sin tratamiento con corticoides pierden la marcha a los 12 años y con tratamiento a los 14-16 años). Actualmente se recomienda el uso de deflazacort a 0.9 mg/k/día y se recomienda su inicio entre los cuatro y cinco años de edad 5.
En los últimos 10 años las enfermedades neuromusculares han experimentado grandes avances en el campo de los ensayos clínicos y la DMD ha sido pionera en estos, tanto en el área de las terapias génicas como en tratar de revertir o bloquear las diferentes consecuencias de la enfermedad.
Estamos en la era de los ensayos clínicos y lo primero es tener adecuadamente fenotipados y genotipados a los pacientes con DMD/B, ya que dependiendo de su estadio clínico (fenotipo) y el tipo de mutación genética (genotipo) se podrá decidir en qué ensayo pueda participar y sobre todo, si los resultados de los ensayos son favorables, se podrá indicar, o no, cada uno de los tratamientos en el futuro (“terapia a la carta”).
Estrategias terapéuticas en distrofia muscular de Duchenne
Con relación a los avances terapéuticos (ensayos clínicos y nuevas terapias), desde un punto de vista didáctico podríamos dividirlas en dos grandes grupos 6:
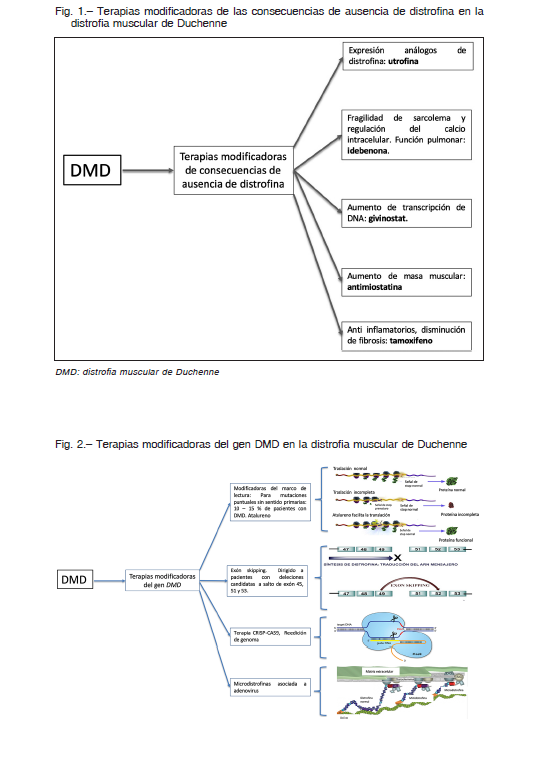
1. Las que actúan sobre los mecanismos fisiopatológicos comunes asociados a la distrofia muscular y que en general no dependen del tipo de mutación: inflamación, capacidad de regeneración muscular, estabilización de la membrana muscular, expresión de proteínas compensatorias como la utrofina, aumento del flujo sanguíneo, antioxidantes, etc. Dado que este tipo de ensayos clínicos y posibles terapias no depende del tipo de mutación, todos los pacientes con DMD podrían ser candidatos a estas terapias (según el estadio clínico de los mismos) (Fig. 1). Actualmente no hay ningún fármaco aprobado, todos están en fase de investigación, a excepción de los corticoides 5, ya comentados en la introducción del artículo.
2. Un segundo grupo de terapias son las llamadas “terapias a la carta” o medicina personalizada. En este
caso el tipo de mutación genética de cada paciente será lo que defina su posible indicación. Como ejemplos de estas terapias tenemos: exón skipping, microdistrofinas, terapia de restauración del marco de lectura, etc. (Fig. 2).
A continuación se comentará de forma breve las principales características de las terapias relacionadas con modificación genética y/o modificación de la expresión de distrofina.
Terapia modificadora del marco de lectura. Atalureno
El atalureno es un compuesto cuyo mecanismo de acción es restaurar el marco de lectura del ARN mensajero, modificando el codón stop prematuro y de esta manera permitir que se continúe con la codificación de la distrofina.
Actualmente el atalureno está autorizado por la EMA (Agencia Europea de Medicamentos) de forma condicional para el tratamiento de pacientes con DMD debida a una mutación sin sentido a partir de los 2 años de edad en ambulantes. Por su mecanismo de acción está indicado en pacientes con DMD que tengan como defecto genético una mutación puntual sin sentido primaria (no secundaria a una microdeleción o microduplicación previa). Los múltiples ensayos clínicos sobre atalureno han demostrado que los pacientes que caminen entre 350 y 450 metros en el test de los seis minutos (6MWT en inglés) y que presenten edades menores de 12 años, se benefician de una prolongación de la marcha (relentización de pérdida de metros en el test de los seis minutos) respecto al grupo control no tratado 7-10.
Microdistrofinas
La terapia de reemplazo de genes se considera una estrategia potencial para el tratamiento de la DMD, con
el objetivo de restaurar la proteína faltante. La terapia génica para la DMD implica introducir un gen de longitud funcionalmente truncado (ADNc) en un vector viral que produzca minidistrofinas que desarrollen una función similar a la distrofina total. El gran tamaño de la transcripción del gen DMD ha sido un obstáculo importante en el desarrollo de métodos para la terapia génica de la DMD. Sin embargo, numerosos estudios tanto en modelos animales como en la clínica, generaron un conocimiento considerable con respecto a los dominios estructurales de la distrofina y permitieron el diseño racional de mini y microdistrofinas altamente funcionales más susceptibles a las aplicaciones de terapia génica (Fig. 2).
La microdistrofina es una versión abreviada de la distrofina, pero conserva sus componentes y funciones principales. Su administración en modelos animales a través de adenovirus demostró la reversión de la sintomatología clínica y los cambios distróficos en la fibra muscular, así como una reducción considerable de los niveles séricos de CPK. A partir de estos resultados favorables, se iniciaran algunos ensayos multicéntricos para comprobar la eficacia de la terapia génica relacionada con las microdistrofinas utilizando como vector los adenovirus 11-15.
Terapia de salto del exón en la distrofia muscular de Duchenne
El principio básico para la terapia del salto del exón es unir un exón previo y uno posterior a la deleción del gen y por consecuencia continuar con el marco de lectura y por tanto con la expresión de distrofina. Los componentes químicos utilizados para tal fin son los oligonucleótidos que son ácidos nucleicos cortos (~ 20 bases) sintéticos y están diseñados para hibridar con secuencias de pre-ARNm complementarias. Actualmente hay varios ensayos clínicos en marcha (salto del exón 51, 53, 45).
A continuación se detallan los ensayos clínicos actuales y las deleciones que se benefician para la terapia del salto del exón:
1. Salto del exón 51, ETEPLIRSEN: (aprobado por FDA para EE.UU.). Deleciones del gen DMD que se benefician del salto del exón 51: 13-50, 29-50,43-50, 45-50, 49-50, 50.
2. Salto del exón 45, CASIMERSEN: Deleciones del gen DMD que se benefician del salto del exón 45: 12-44, 18-44, 44, 46-47, 46-48, 46-49, 46-51, 46-53, 46-55.
3. Salto del exón 53, GOLODIRSEN: Deleciones del gen DMD que se benefician del salto del exón 45, 42-52, 45-52, 47-52, 48-52, 49-52, 50-52, 52, 54-58.
Algunos de los inconvenientes de la terapia de salto del exón con oligonucleótidos antisentido es la corta eficacia (por lo que es necesario su administración de forma regular) y mala captación en órganos vitales como el corazón 16-18.
Salto de exón a través de la edición del genoma: terapia CRISPR/Cas9
Como se mencionó anteriormente, los oligonucleótidos antisentido presentan algunos inconvenientes: una mala captación en órganos vitales como el corazón y una eficacia de corta duración. Para superar estos problemas, la edición de genes asociada a repeticiones palindrómicas cortas (CRISPR/Cas9), es una terapia prometedora con la cual se logra eliminar de forma permanente la mutación-deleción del gen y se logra restaurar el marco de lectura de la proteína. Los vectores AAV (serotipo 8 o 9) se han utilizado para administrar los componentes de CRISPR/Cas9 in vivo, ya sea por vía intramuscular, intraperitoneal o intravenosa para eliminar el exón mutado 23 del gen DMD del genoma de ratones mdx, estos estudios mostraron distrofina restaurada, tanto en los músculos esqueléticos como cardíacos, confirmados por inmunotinción y Western blot. La tecnología CRISPR/Cas9 produce esencialmente los mismos resultados que la omisión de exón con oligonucleótidos antisentido, pero con la ventaja de inducir correcciones permanentes del gen DMD mutado sin requerir tratamiento repetitivo.
Comentarios finales sobre estrategias terapéuticas en distrofia muscular de Duchenne
Los avances actuales y de futuro en el campo de las distrofias musculares son el reflejo de la transformación de enfermedades con poca esperanza de vida y solo tratamiento sintomático en enfermedades crónicas con una mejor calidad y tiempo de supervivencia y, en algunos casos, revertir el proceso fisiopatológico y curar la enfermedad. El tratamiento fundamental para la DMD es la suplementación o recuperación de distrofina y el uso de vectores virales. La terapia del salto del exón o terapias que modifiquen el marco lectura, como se describen en esta revisión, tienen como objetivo común la producción
de distrofina, y por tanto mejorar o revertir los síntomas de la enfermedad dependiendo del estadio clínico de la misma 19, 20.
Es probable que un tratamiento eficaz para la DMD requiera combinaciones de terapias que se ocupen tanto del defecto primario como sus consecuencias fisiopatológicas secundarias. Mientras se llega a la cura definitiva de la DMD, es fundamental que los pacientes y sus familias reciban la atención basada en los guías médicas del cuidado integral en cada uno de los períodos de la enfermedad.
Conflicto de intereses: Ninguno para declarar
Bibliografía
1. Birnkrant, D, Bushby K, Bann CM, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol 2018; 17: 251-67.
2. Birnkrant, D, Bushby K, Bann CM, et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol 2018; 17: 347-61.
3. Birnkrant, D, Bushby K, Bann CM, et al. Diagnosis and management of Duchenne muscular dystrophy, part 3: primary care, emergency management, psychosocial care, and transitions of care across the lifespan. Lancet Neurol 2018; 17: 445-55.
4. Noritz G, Naprawa J, Apkon SD, et al. Primary care and emergency department management of the patient with Duchenne muscular dystrophy. Pediatrics 2018; 142 (Suppl 2): S90-8.
5. McDonald CM, Henricson EK, Abresch RT, et al. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: a prospective cohort study. Lancet 2018; 391: 451-61.
6. Verhaart IEC, Aartsma-Rus A. Therapeutic developments for Duchenne muscular dystrophy. Nat Rev Neurol 2019; 15: 373-86.
7. McDonald CM, Campbell C, Torricelli RE, et al. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017; 390: 1489-98.
8. Korinthenberg R. A new era in the management of Duchenne muscular dystrophy. Dev Med Child Neurol 2019; 61: 292-7.
9. Vita G, Vita GL, Musumeci O, Rodolico C, Messina S. Genetic neuromuscular disorders: living the era of a therapeutic revolution. Part 2: diseases of motor neuron and skeletal muscle. Neurol Sci 2019; 40: 671-81.
10. Landfeldt E, Sejersen T, Tulinius M. A mini-review and implementation model for using ataluren to treat nonsense mutation Duchenne muscular dystrophy. Acta Paediatr 2019; 108: 224-30.
11. Ho PP, Lahey LJ, Mourkioti F, et al. Engineered DNA plasmid reduces immunity to dystrophin while improving muscle force in a model of gene therapy of Duchenne dystrophy. Proc Natl Acad Sci U S A 2018; 115: E9182-91.
12. Filareto A, Maguire-Nguyen K, Gan Q, et al. Monitoring disease activity noninvasively in the mdx model of Duchenne muscular dystrophy. Proc Natl Acad Sci U S A 2018; 115: 7741-6.
13. Delalande O, Molza AE, Dos Santos Morais R, et al. Dystrophin’s central domain forms a complex filament that becomes disorganized by in-frame deletions. J Biol Chem 2018; 293: 6637-46.
14. Halbert CL, Allen JM, Chamberlain JS. AAV6 vector production and purification for muscle gene therapy. Methods Mol Biol 2018; 1687: 257-66.
15. Le Guiner C, Servais L, Montus M, et al. Long-term microdystrophin gene therapy is effective in a canine model of Duchenne muscular dystrophy. Nat Commun 2017; 8: 16105.
16. Khan N, Eliopoulos H, Han L, et al. Eteplirsen treatment attenuates respiratory decline in ambulatory and nonambulatory patients with Duchenne muscular dystrophy. J Neuromuscul Dis 2019; 6: 213-25.
17. Watanabe N, Nagata T, Satou Y, et al. NS-065/NCNP-01: an antisense oligonucleotide for potential treatment of exon 53 skipping in Duchenne muscular dystrophy. Mol Ther Nucleic Acids 2018; 13: 442-9.
18. Li D, Mastaglia FL, Fletcher S, Wilton SD. Precision medicine through antisense oligonucleotide-dediated exon skipping. Trends Pharmacol Sci 2018; 39: 982-94.
19. Cai A, Kong X. Development of CRISPR-mediated systems in the study of Duchenne muscular dystrophy. Hum Gene Ther Methods 2019; 30: 71-80.
20. Lim QRK, Yoon C, Yokota T. Applications of CRISPR/Cas9 for the treatment of Duchenne muscular dystrophy. J Pers Med 2018; 8. pii: E38.