
CATALINA M. UNGARO 1, 2, M. SILVINA ODSTRCIL-BOBILLO 1, PAULA M. RUSSO 1, 2
1 Departamento de Medicina Interna, 2 Área de investigación en Medicina Interna,
Hospital Italiano de Buenos Aires, Argentina
Resumen El síndrome de Gitelman forma parte de las denominadas tubulopatías perdedoras de sal. El bloqueo parcial de la reabsorción de sodio en el túbulo contorneado distal determina la aparición de hipokalemia e hipomagnesemia. Se realizó un estudio descriptivo de una serie de cinco casos de síndrome de Gitelman (4 mujeres, de 28 a 85 años de edad) atendidos en nuestra institución entre los años 2004 y 2015. La forma de diagnóstico más frecuente en nuestra serie fue por hallazgo de laboratorio. El único síntoma clínico manifestado en forma espontánea fue astenia. En cuanto a los valores de laboratorio, la potasemia fue 2.5 ± 0.5 mmol/l, con un valor mínimo de 2.1. Adicionalmente, el valor de magnesio en sangre fue 1.3 ± 0.3 mg/dl. Como conclusión, observamos que las formas de presentación consisten en alteraciones bioquímicas con o sin manifestaciones inespecíficas, lo que representa actualmente la mayor dificultad diagnóstica y refuerza la importancia de lograr un diagnóstico oportuno, en especial en pacientes jóvenes y con valores críticos de potasio sérico.
Palabras clave: síndrome de Gitelman, hipopotasemia, hipomagnesemia
Abstract Gitelman syndrome is one of the salt losing tubulopathies. Hypokalemia and hypomagnesemia appear in the setting of the partial blockade of salt absorption in the distal tubule. We conducted a descriptive study of a case series of five patients with Gitelman syndrome (4 women, from 28 to 85 years) in our institution, between the years 2004 and 2015. The most frequent form of diagnosis in our series was by laboratory finding. The only acknowledged clinical symptom was malaise. Regarding laboratory findings, the mean potassemia was of 2.5 ± 0.5 mmol/l, with a minimum value of 2.1 mmol/l. Additionally, the serum magnesium value was of 1.3 ± 0.3 mg/dl. In conclusion, we observed that the forms of presentation consist of biochemical alterations with or without nonspecific manifestations, which currently represents the greatest diagnostic difficulty and reinforces the importance to achieve a timely diagnosis, especially in young patients with critical serum potassium values.
Key words: Gitelman syndrome, hypokalemia, hypomagnesemia
Dirección postal: Catalina Ungaro, Hospital Italiano de Buenos Aires, Tte. Gral. J. D. Perón 4190, 1199 Buenos Aires, Argentina
e-mail: catalina.ungaro@hospitalitaliano.org.ar
El síndrome de Gitelman forma parte de las denominadas tubulopatías perdedoras de sal 1, 2. Dentro de este grupo de enfermedades se encuentran el síndrome de Bartter, que afecta el asa gruesa de Henle, así como el síndrome de Gitelman, donde el compromiso reside en el túbulo contorneado distal 3. Se estima que la prevalencia de este síndrome es de 1/40 000, constituyendo uno de los desórdenes tubulares renales hereditarios más frecuentes 4. Las manifestaciones clínicas de ambos síndromes dependen de las consecuencias del bloqueo parcial de la función de estos segmentos y su implicancia en la homeostasis del agua y electrolitos. En el caso del síndrome de Gitelman, la presencia del cotransportador de sodio-cloro no funcionante en el túbulo contorneado distal, debido a mutaciones en el gen que lo codifica, determina una pequeña pérdida de cloruro de sodio, ya que la mayoría se ha reabsorbido en segmentos más proximales. Sin embargo, se genera una pérdida distal de potasio y protones por el aumento de la oferta distal de sodio 5-7. El balance del magnesio y el calcio también se ven afectados, completando el cuadro la hipomagnesemia e hipocalciuria 8, 9.
Dada la baja frecuencia de esta enfermedad y a lo inespecífico de su presentación clínica, es frecuente que existan fallas o retrasos en el diagnóstico 10 y es importante estar alerta para realizarlo en forma oportuna. Es por esto que se presentan cinco casos con diagnóstico de síndrome de Gitelman observados en un hospital de tercer nivel de la ciudad de Buenos Aires, en el período de 2004-2015. Se describen las características clínicas y de laboratorio, así como la evolución de la enfermedad.
Caso clínico 1
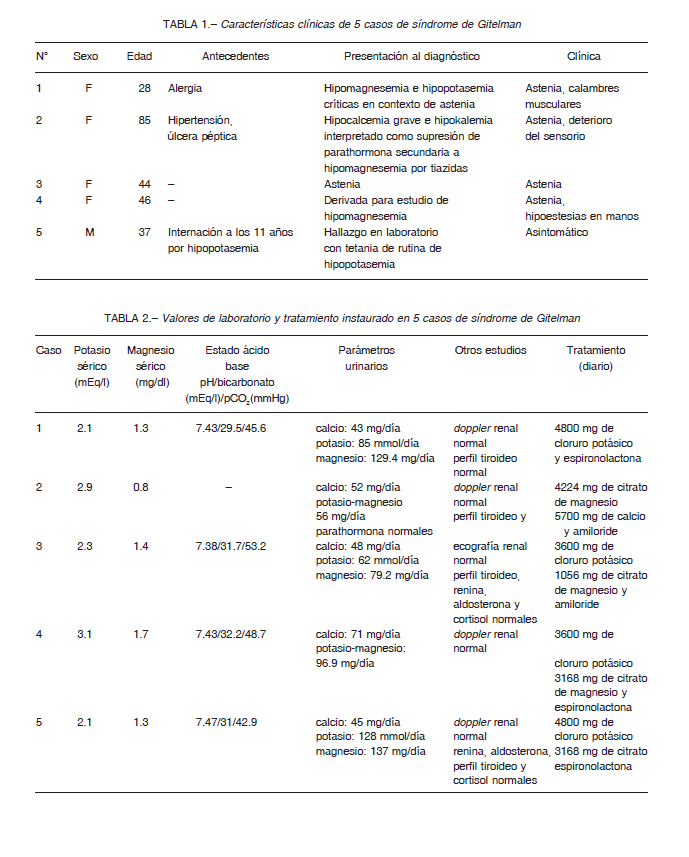
Mujer de 28 años, sin antecedentes clínicos de relevancia más allá que alergias leves tratadas ocasionalmente con antihistamínicos. Se encontraba en seguimiento para estudio de un rash inespecífico cuando se halló una kalemia de 2.1 mmol/l asociado a hipomagnesemia de 1.3 mgl/dl y alcalosis metabólica en el análisis de laboratorio. Al interrogatorio dirigido realizado por el médico que la evaluó, se encontró únicamente astenia como síntoma, asociado a calambres musculares esporádicos. El examen físico no presentó particularidades y la presión arterial era normal.
Se realizaron mediciones de excreción de iones en orina con hallazgo de hipocalciuria y excreción aumentada de potasio y magnesio. Otros estudios complementarios incluyeron un doppler renal y un perfil hormonal tiroideo que resultaron normales. Se llegó al diagnóstico de síndrome de Gitelman y como tratamiento inicial se instauró la reposición vía oral de potasio y magnesio, hubo una evolución tórpida que requirió cuatro internaciones para reposición electrolítica endovenosa, sin otras intercurrencias o complicaciones. Finalmente, se logró estabilidad clínica y valores iónicos séricos normales con el agregado de espironolactona.
Caso clínico 2
Mujer de 85 años, hipertensa y con antecedentes de úlcera péptica. Había sido internada recientemente por hipocalcemia grave (4.5 mg/dl), hipomagnesemia (0.8 mg/dl) e hipokalemia (2.8 mmol/l) interpretadas como supresión de parathormona secundaria a hipomagnesemia crónica por consumo de tiazidas y omeprazol. Al momento de la internación, refirió astenia y mostró deterioro del sensorio.
Durante la internación evolucionó con alteraciones en algunos de los análisis de laboratorio, persistentes a pesar de la suspensión de ambos fármacos y la adecuada reposición electrolítica.
Se realizaron como exámenes complementarios un doppler renal y un perfil hormonal tiroideo y paratiroideo que resultaron normales, llegándose al diagnóstico de síndrome de Gitelman.
El tratamiento al alta, luego de normalizados los valores de laboratorio, incluyó reposición de magnesio y potasio con el agregado de amiloride.
Caso clínico 3
Mujer de 44 años, sin antecedentes clínicos de relevancia. Consultó a su médico de cabecera por astenia de larga data. Al momento de la evaluación, el examen físico fue normal al igual que la presión arterial. Se solicitó un examen de laboratorio que reveló hipokalemia (2.3 mmol/l) e hipomagnesemia (1.4 mg/dl) junto con alcalosis metabólica. El estudio urinario mostró hipocalciuria junto con excreción aumentada de potasio y magnesio. Para completar la valoración y descartar diagnósticos diferenciales, se realizó un doppler renal con estudio de los ejes tiroideo (TSH) y suprarrenal (renina, aldosterona y cortisol) que resultaron normales.
Con diagnóstico de síndrome de Gitelman se inició la reposición de iones y amiloride para obtener valores iónicos cercanos a la normalidad.
Caso clínico 4
Mujer de 46 años derivada para estudio de hipomagnesemia, presentaba astenia e hipoestesias en ambas manos. No refería arterial 90/60 mmHg. El laboratorio informó hipokalemia (3.1 mmol/l) e hipomagnesemia (1.7 mg/dl) acompañadas de un trastorno mixto opuesto (alcalosis metabólica y acidosis respiratoria) en el estado ácido base. La excreción urinaria de calcio se encontró reducida mientras que la de potasio y magnesio aumentadas. Se realizó en forma complementaria un doppler renal y una determinación de TSH sérica que resultaron normales. El tratamiento consistió en la reposición oral de potasio y magnesio más el uso de espironolactona, de esta manera logró mantener valores normales de magnesio y cercanos a la normalidad de potasio, con mejoría sintomática.
Caso clínico 5
Hombre de 35 años. Había presentado a los 11 años una internación en otro centro por hipokalemia grave con tetania en contexto de diarrea. Ingresó a nuestro centro con un hallazgo de hipokalemia (2.7 mmol/l) en un análisis de rutina. Al interrogatorio dirigido refirió presentar contracturas (junto con signo de Trousseau) de larga data e hipoacusia. Al momento de la consulta se encontraba asintomático, el examen físico fue normal y la presión arterial de 100/60 mmHg. El ECG reveló T aplanadas. Un nuevo examen de laboratorio mostró hipokalemia (2.1 mmol/l) e hipomagnesemia (1.3 mg/dl) junto con alcalosis metabólica. El análisis de orina reveló hipocalciuria con excreciones de potasio y magnesio aumentadas.
Se completó el estudio con TSH, renina, aldosterona y cortisol séricos normales. Para confirmación de síndrome de Gitelman se realizó el test con tiazidas que resultó positivo.
Luego de múltiples infusiones endovenosas de potasio y magnesio, se logró iniciar tratamiento vía oral de reposición electrolítica más espironolactona, logrando de esta manera estabilidad clínica y presentando como complicación disminución del deseo sexual por lo que se decidió rotar la espironolactona por amiloride con adecuada respuesta.
Discusión
Se presentan cinco casos de pacientes con diagnóstico de síndrome de Gitelman. A pesar de ser una enfermedad infrecuente, ha adquirido relevancia dado la afectación, en general, de personas jóvenes y con valores críticos de potasio 1, hecho que hemos podido ver en nuestra serie de casos, donde el promedio de edad fue de 48 años (DE 21.9). Sin embargo, a diferencia de lo presentado en la literatura, donde la mayor prevalencia se observa en hombres como consecuencia de resultar más sintomático 8, 10, notamos en nuestro medio una mayor frecuencia en mujeres (4 de 5 casos). Una posible explicación a ello sería la forma de presentación, dado que en todos los casos el diagnóstico fue a partir de análisis pedidos por otra causa.
Al igual que lo descrito en la literatura, los síntomas de presentación en nuestro medio consistieron mayormente en manifestaciones inespecíficas 10,11. El único síntoma que manifestaban en forma espontánea fue astenia y solo uno de los pacientes refirió antecedentes compatibles en la infancia; había sido internado por un cuadro de gastroenteritis por hipokalemia y tetania. Adicionalmente, en dos casos, el interrogatorio dirigido reveló la presencia de calambres e hipoestesias (Tabla 1). En cuanto a los valores de laboratorio, en nuestra serie la kalemia fue 2.5 ± 0.5 mmol/l con un valor mínimo de 2.1 mmol/l y la magnesemia fue 1.3 ± 0.3 mg/dl con un mínimo de 0.8 mg/dl. Los pacientes que presentaban estado ácido base tenían alcalosis metabólica asociada (4 de 5) y todos tenían pérdida aumentada de potasio y magnesio urinario con hipocalciuria, parámetros urinarios compatibles con síndrome de Gitelman (Tabla 2). Ante estos resultados, el médico clínico debe sospechar esta afección para realizar otros análisis, como ionograma y estado ácido base, que no suelen estar incluidos en un examen de rutina. Se refuerza la importancia de los avances científicos en la búsqueda de los responsables genéticos con el fin de promover la detección precoz de esta enfermedad.
El tratamiento instaurado en nuestro medio consistió en la reposición de iones, asimismo se requirió indicación de espironolactona o amiloride, lográndose valores de kalemia cercanos a la normalidad en todos los casos. El magnesio alcanzó un valor promedio de 1.6 mg/dl. Considerando que el objetivo del tratamiento en esta afección reside en la mejoría sintomática 5, la que se logró en todos los casos de nuestra serie, la resolución de los cuadros es altamente factible.
Finalmente, es de importancia resaltar que luego del diagnóstico, dos de los cinco pacientes requirieron internación para estabilización de los valores iónicos en forma endovenosa, siendo el promedio de días de internación de 1.1 días, lo que marca la potencial gravedad de éste desorden.
Podemos concluir que se trata de una enfermedad rara, en pacientes jóvenes, cuyos síntomas iniciales son inespecíficos y la forma de llegar al diagnóstico, más frecuente, es como hallazgo de laboratorio. Sin embargo, puede desencadenar complicaciones potencialmente graves. Por todo esto es fundamental entrenar a médicos para reconocer la afección y permanecer alertas con el objetivo de realizar diagnóstico oportuno del síndrome, dado que con la reposición electrolítica y el complemento farmacológico adecuado es posible que los afectados logren valores iónicos cercanos a la normalidad y alcancen la mejoría sintomática.
Conflicto de intereses: Ninguno para declarar
Bibliografía
1. Seyberth HW, Schlingmann KP. Bartter- and Gitelman-like syndromes: salt-losing tubulopathies with loop or DCT defects. Pediatr Nephrol 2011; 26: 1789-802.
2. Zelikovic I. Hypokalaemic salt-losing tubulopathies: anevolving story. Nephrol Dial Transplant 2003; 18: 1696-700.
3. Wittner M, Di Stefano A, Wangemann P, Greger R. How do loop diuretics act? Drugs 1991; 41 Suppl 3: 1-13.
4. Knoers NV, Levtchenko EN. Gitelman syndrome. Orphanet J Rare Dis 2008; 3: 22.
5. Blanchard A, Bockenhauer D, Bolignano D, et al. Gitelman syndrome: consensus and guidance from a kidney disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int 2017; 91: 24-33.
6. Brunton L, Chabner B, Knollman B. Goodman and Gilman’s The Pharmacological Basis of Therapeutics, 12th
ed; 2011, p 1808.
7. Ardalan M, Golzari SEJ. An integrated view of potassium homeostasis. N Engl J Med 2015; 373: 1787.
8. Cruz DN, Shaer AJ, Bia MJ, et al. Gitelman’s syndrome revisited: An evaluation of symptoms and health-related quality of life. Kidney Int 2001; 59: 710-7.
9. Cole DE, Quamme GA. Inherited disorders of renal magnesium handling. J Am Soc Nephrol. 2000; 11: 1937-47.
10. Fujimura J, Nozu K, Yamamura T, et al. Clinical and genetic characteristics in patients with gitelman syndrome. Kidney Int Rep 2018; 4: 119-25.
11. Vidal Company A, Ruiz Cano R, Gutiérrez Junquera C, Lillo Lillo M, Onsurbe Ramírez I. Variabilidad fenotípica del síndrome de Gitelman. An Esp Pediatr 2000; 52: 285-8.