
VANINA VACCHIANO, SILVIA SELEME, ADRIÁN F. DALY, ALBERT BECKERS, HERNÁN VALDÉS-SOCIN
Department of Endocrinology, Centre Hospitalier Universitaire de Liège, University of Liege, Liege, Belgium
Resumen La mayoría de los adenomas hipofisarios son esporádicos, pero un 3-5% puede ocurrir en un contexto familiar y hereditario. Este es el caso de la neoplasia endocrina múltiple de tipo 1 (NEM1), complejo de Carney (CNC) y adenomas hipofisarios aislados familiares (FIPA). El FIPA es una condición infrecuente, que ocurre en un contexto familiar, no asociada a NEM t ipo1 ni CNC. Los FIPA pueden ser homogéneos (todos los adenomas tienen el mismo fenotipo) o heterogéneos (diferente fenotipo tumoral). Describimos una familia congolesa en la que dos hermanas y una prima fueron diagnosticadas a los 29, 32 y 40 años, respectivamente, con un prolactinoma (FIPA homogéneo). Las pacientes presentaron macroadenomas no invasivos al momento del diagnóstico, con buena respuesta biológica y tumoral al tratamiento con cabergolina hasta una dosis máxima de 1.5 mg/semanal. De las dos hermanas, una cursó un embarazo sin complicaciones. Durante el seguimiento de 12 años, ninguna de ellas presentó elementos clínicos o biológicos compatibles con NEM1 o CNC, por lo que dichos genes no se estudiaron. El análisis genético en dos de las pacientes permitió descartar la posibilidad de una mutación germinal del gen aryl hydrocarbon receptor interacting protein (AIP). Se considera que el 80% de los pacientes con FIPA no presentan mutación del gen AIP, por lo que se requieren futuros estudios en este tipo de familias, para poder determinar otros genes afectados involucrados en su fisiopatología.
Palabras clave: FIPA, AIP, prolactinoma familiar, embarazo, cabergolina
Abstract Most pituitary adenomas are sporadic, but 3-5% can occur in a family and hereditary context. This is the case of multiple endocrine neoplasia type 1 (MEN1), Carney complex (CNC) and familial isolated pituitary adenomas (FIPA). FIPA is an infrequent condition that occurs in a family context, not associated with MEN type1 or CNC. FIPA kindred can be homogeneous (all adenomas affected in the family having the same tumor phenotype) or heterogeneous (different tumor phenotypes in the affected members). We describe a Congolese family in which two sisters and a cousin were diagnosed with a prolactinoma (homogenous FIPA) at the ages of 29, 32 and 40 years, respectively. The patients presented with macroadenomas at the time of diagnosis, non-invasive tumors and good biological response to cabergoline treatment (maximum dose of 1.5 mg/weekly). Of these two sisters, one went through a pregnancy without complications. Because no MEN1 and CNC clinical and biochemical features were detected during the 12-year follow-up, these genes were not investigated. The genetic analysis of the aryl hydrocarbon receptor interacting protein (AIP) was normal. As nearly 80% of patients with FIPA do not have a mutation in the AIP gene, future studies in these families are required to identify other affected genes involved in their physiopathology.
Key words: FIPA, AIP, familial prolactinoma, pregnancy, cabergoline
Dirección postal: Hernán Valdés-Socin, Servicio de Endocrinología, Centre Hospitalier Universitaire de Liège, Rue de Gaillarmont 600, 4030 Liège, Bélgica
e-mail: hg.valdessocin@chuliege.be
Los adenomas pituitarios clínicamente aparentes se presentan en 1:1000 personas en Europa 1, de éstos aproximadamente solo el 3-5% son familiares. La neoplasia endócrina múltiple tipo 1 (NEM1) y el complejo de Carney (CNC) son enfermedades hereditarias que se asocian a adenomas pituitarios 2. Entre 2000-2006 hemos identificado y caracterizado una nueva enfermedad en familias afectadas con adenomas hipofisarios aislados no relacionados con NEM1/CNC 3. Acuñamos entonces el término familial isolated pituitary adenomas (FIPA) para definirlas, empleando la denominación homogénea para describir familias con un mismo fenotipo y heterogéneo cuando los fenotipos tumorales son distintos 4. Casi dos décadas después, varios cientos de familias FIPA han sido identificadas.
En la observación de los FIPA, se han registrado todos los tipos de adenomas, tanto funcionantes cómo no
funcionantes. Dentro de la presentación homogénea, se presenta acromegalia en 58%, prolactinoma en 32%, no funcionantes en 7% y raramente casos de enfermedad de Cushing. Por otro lado, del grupo heterogéneo, el fenotipo más frecuente es la combinación de acromegalia con prolactinomas (42%) 5. En la mayoría de los pacientes, alrededor del 75%, existe una relación de primer grado entre los afectados 6. En el 2006, se describió la asociación de la mutación del gen aryl hydrocarbon receptor interacting protein (AIP) en la patogénesis de los adenomas pituitarios familiares, a partir de lo cual se realizaron múltiples estudios, tanto en la población general como en grupo específicos 6, 7. El gen AIP, situado en el cromosoma 11q13.3, contiene 6 exones y codifica para una proteína de 330 aminoácidos que se expresa variablemente en los tejidos 6, 7. Dentro de los tumores hipofisarios aislados familiares, se han descripto mutaciones en el gen AIP, principalmente en familias FIPA con acromegalia homogénea o con acromegalia asociada a prolactinoma; en individuos con acrogigantismo (29% de los casos) y pacientes jóvenes (<18 años) con macroadenomas esporádicos 8.
En familias con gigantismo, con hipersecreción de GH y prolactina asociada, es importante descartar el síndrome de acrogigantismo ligado al cromosoma X, causado por una micro duplicación en Xq26.3 que comprende el gen orphan G-protein coupled receptor (GPR101) 9, 10.
La mutación del AIP explica solo una minoría (aproximadamente el 20%) de las familias con FIPA 6. Múltiples estudios avalan que los tumores con mutaciones en AIP son diagnosticados a edades más tempranas (78% menores de 30 años), con predominio en el sexo masculino (61%), son más agresivos y de mayor tamaño (88%), en comparación con aquellos sin mutaciones en dicho gen 6. Además, los tumores en pacientes con AIP mutado presentan principalmente resistencia al tratamiento farmacológico (con agonistas dopaminérgicos y análogos de la somatostatina) en comparación con aquellos sujetos sin mutaciones en AIP 11. En este trabajo se describe una familia FIPA con tres afectados que presentan prolactinomas, no relacionados con NEM1 ni CNC, y en los que el estudio genético en AIP resultó negativo en dos de ellos.
Caso clínico 1
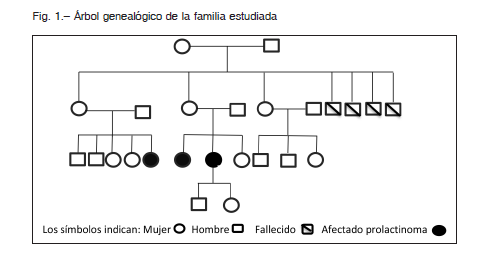
Mujer de 40 años con antecedentes personales de infección por el virus de inmunodeficiencia humana (HIV) tratada con terapia antirretroviral. Consultó por primera vez en el año 2006 al servicio de endocrinología del Hospital de Lieja por amenorrea e hiperprolactinemia. Refirió macroprolactinoma en tratamiento con cabergolina 0.5 mg semanal, con mala adherencia al tratamiento. Mencionó antecedentes familiares en una hermana con diagnóstico de macroprolactinoma, con igual plan terapéutico (Fig. 1). Se constató una hiperprolactinemia de 1733 mU/l (valor normal: 100-400 mUI/l) como único hallazgo patológico en el laboratorio. La RMN de hipófisis realizada el 09/11/2001 evidenció un nódulo adenomatoso intraselar de 11 × 10 × 9 mm. Se decidió aumentar la dosis de cabergolina a 0.75 mg/semanal. Al término de tres años, evolucionó con descenso de los valores de prolactina (100 mUI/l en 2009) y desaparición radiológica de la imagen de adenoma hipofisario. Se disminuyó entonces la dosis de cabergolina a 0.50 mg/semanal. El análisis del gen AIP se realizó extrayéndose el ADN de linfocitos periféricos y la búsqueda de mutaciones se efectuó por secuenciación directa de acuerdo con la metodología que describimos en trabajos anteriores 5, 7, no detectándose ninguna mutación.
Caso clínico 2
Mujer de 32 años, hermana del caso 1 (Fig. 1). Presentó antecedentes de macroprolactinoma, diagnosticado en el 2001, en tratamiento con cabergolina 1.5 mg/semanal. Acudió al servicio de endocrinología por primera vez el 28/11/2006. Refirió cefaleas frecuentes a predominio derecho, con ciclos menstruales irregulares (oligomenorrea) al suspender la medicación.
La RMN de hipófisis en el 2001 reveló un adenoma hipofisario de 11 × 11 × 12 mm, que deformaba el piso de la silla turca. Debido a la mala adherencia al tratamiento y a los controles médicos, no se logró la normalización de los valores de prolactina (3200 mUI/l al momento del diagnóstico y 650 mUI/l durante el tratamiento con cabergolina). A pesar de esto, la paciente cursó un embarazo en el 2008, intercurriendo con diabetes gestacional, con producto normal.
Caso clínico 3
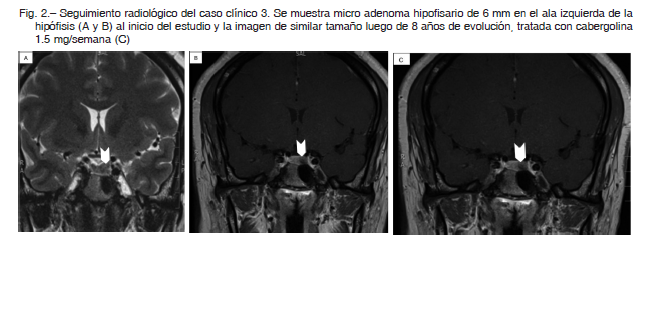
Mujer de 29 años, prima de casos 1 y 2 (Fig. 1) con antecedentes de bocio multinodular en tratamiento con levotiroxina. Consultó por primera vez el 27/04/2009 por galactorrea de cinco años de evolución, acompañada de amenorrea en los últimos nueve meses. Refirió además cefaleas frontales y visión borrosa. En la ecografía tiroidea se observaba un nódulo de 2.6 cm. Su nivel de prolactina era de 3000 mU/l. La RMN de hipófisis del 12/06/2009 mostraba un microadenoma hipofisario de 6 mm de diámetro lateralizado a la izquierda (Fig. 2). Se inició tratamiento farmacológico con cabergolina 0.5 mg/semanal. Se realizó tiroidectomía total en el mismo año por bocio multinodular, con anatomía patológica benigna. El estudio genético excluyó mutaciones en el gen AIP. A pesar de la toma regular de la medicación, la paciente persist ía con altos valores de TSH: 6 mUI/l (valores normales: 0.3-4 mUI/l) y prolactina 800 mU/l, por lo que requirió aumento de la dosis de levotiroxina (150 μg/día) y cabergolina (1.5 mg/semanal) hasta normalizar dichos parámetros. La RMN de hipófisis del 07/03/2017 mostraba una imagen de adenoma intraselar de tamaño estable (Fig. 2).
Durante el seguimiento de las tres pacientes no se constató ningún signo clínico, tumoral ni alteraciones bioquímicas relacionadas con NEM1 ni CNC.
Discusión
Los prolactinomas son los tumores pituitarios secretores de hormonas más frecuentes, representando aproximadamente el 40% de todos los adenomas hipofisarios. Con mayor prevalencia entre 20 a 50 años de edad, existe una amplia preponderancia en el sexo femenino (mujer/hombre 10:1). Esta diferencia se iguala en el grupo etario mayor a 50 años 12. En este informe describimos una familia de tres mujeres afectadas por prolactinomas, en ausencia de signos clínicos de NEM tipo 1 y CNC.
Dada la baja prevalencia de los adenomas esporádicos (1/1000) 1, estimamos que la probabilidad de presentar tres prolactinomas en el mismo núcleo familiar, es muy reducida. Las mutaciones en AIP explican solo una minoría de los casos de FIPA, y se la encuentra con más frecuencia en familias con acromegalia homogénea, o heterogénea con acromegalia y prolactinoma.
Sus afectados suelen ser diagnosticados a edades más tempranas y su respuesta al tratamiento farmacológico
es irregular, aunque se ha descrito la remisión clínica en un caso 13. A su vez, las familias sin mutaciones en AIP suelen presentar frecuentemente prolactinomas 5-7, que al momento del diagnóstico son microadenomas como los casos esporádicos, y tienen asimismo respuesta favorable a la terapéutica 5, 6.
Si bien dos de las pacientes mostraron adenomas mayores a 1cm, todas tuvieron buena respuesta al tratamiento con cabergolina, logrando la normalización de los parámetros hormonales, con favorable evolución radiológica. En dos de los casos, hubo desaparición radiológica de la lesión hipofisaria luego del tratamiento.
El tratamiento con cabergolina permitió, inclusive, un embarazo en una de las pacientes.
En función de la literatura y de nuestra experiencia, enfatizamos la importancia de realizar un exhaustivo interrogatorio y un árbol genealógico ante un paciente que presente un adenoma pituitario, en busca de otros miembros afectados, para descartar que se trate de un adenoma pituitario aislado familiar 5. En presencia de tumores hipofisarios diagnosticados en edades tempranas, sobre todo niños y adolescentes con somatotropinomas y prolactinomas, se debe considerar la búsqueda de mutaciones en el gen AIP. También deben considerarse otras causas genéticas asociadas a adenomas pituitarios como NEM tipo1, complejo de Carney y mutaciones en el gen Cyclin-dependent kinase 1 inhibitor B (CDKN1B), entre otras 5. A lo largo de 12 años de seguimiento, no se evidenció crecimiento tumoral ni otras anomalías bioquímicas sugestivas de estos tres síndromes en nuestros pacientes. No obstante, hacemos notar que exceptuando el gen AIP, dichos genes no fueron investigados en la presente comunicación.
En relación al gen AIP, la portación de mutaciones es alrededor del 20% en las familias estudiadas 5, 7, 9, 14 y, si bien no existe consenso sobre cuál es el mejor seguimiento de los pacientes con mutación AIP, ante casos asintomáticos, parece prudente realizar análisis de sangre anuales (cómo mínimo IGF-1 y prolactina) y RMN de hipófisis en caso de hallarse anormalidades bioquímicas, síntomas o signos de presencia tumoral 5. Siguen siendo necesarios más estudios para poder identificar otras mutaciones causales de FIPA.
Agradecimientos: Este trabajo fue concebido en el marco del convenio de cooperación científica entre la
Facultad de Medicina de la Universidad de Medicina de Buenos Aires y la Universidad de Lieja de Bélgica, firmado el 22.12.2015.
Conflicto de intereses: Ninguno para declarar
Bibliografía
1. Daly AF, Rixhon M, Adam C, Dempegioti A, Tichomirowa MA, Beckers A. High prevalence of pituitary adenomas: a cross-sectional study in the province of Liege, Belgium. J Clin Endocrinol Metab 2006; 91: 4769-75.
2. Beckers A, Daly AF. The clinical, pathological and genetic features of familial isolated pituitary adenomas. Eur J Endocrinol 2007; 157: 371-82.
3. Valdés-Socin H, Poncin J, Stevens V, Stevenaert A, Beckers A. Adénomes hypophysaires familiaux isoles non liés avec la mutation somatique NEM-1. Suivi de 27 patients. En : htpp://hdl.handle.net/2268 /64787; consultado septiembre 2019.
4. Valdés-Socin H, Poncin J, Vambelinghen JF, et al. Familial pituitary tumours. En: Tamburrano G, Casanueva F, Baldelli R, eds. Dalla Riserca di base alla Clinica. Pubblicazioni Medici Scientifiche LILLY, 2001, p 24-28.
5. Daly AF, Beckers A. Familial isolated pituitary adenomas (FIPA) and mutations in the aryl hydrocarbon receptor interacting protein (AIP) gene. Endocrinol Metab Clin North Am 2014; 4: 19-25.
6. Beckers A, Aaltonen LA, Daly AF, Karhu A. Familial isolated pituitary adenomas (FIPA) and the pituitary adenoma predisposition due to mutations in the aryl hydrocarbon receptor interacting protein (AIP) gene. Endocr Rev 2013; 34: 239-77.
7. Daly AF, Vanbellinghen JF, Khoo SK, et al. Aryl hydrocarbon receptor-interacting protein gene mutations in familial isolated pituitary adenomas: analysis in 73 families. J Clin Endocrinol Metab 2007; 92: 1891-6.
8. Rostomyan L, Daly AF, Petrossians P, et al. Clinical and genetic characterization of pituitary gigantism: an international collaborative study in 208 patients. Endocr Relat Cancer 2015; 22: 745-57.
9. Trivellin G, Daly AF, Faucz FR, et al. Gigantism and acromegaly due to Xq26 microduplications and GPR101 mutation. N Engl J Med 2014; 371: 2363-74.
10. Beckers A, Lodish MB, Trivellin G, et al. X-linked acrogigantism syndrome: clinical profile and therapeutic responses. Endocr Relat Cancer 2015; 22: 353-67.
11. Daly A, Tichomirowa MA, Petrossians P, et al. Clinical characteristics and therapeutic responses in patients with germ-line AIP mutations and pituitary adenomas: an international collaborative study. J Clin Endocrinol Metab 2010; 95: E373-83.
12. Vilar L, Abucham J, Albuquerque JL, et al. Controversial issues in the management of hyperprolactinemia and prolactinomas – An overview by the Neuroendocrinology Department of the Brazilian Society of Endocrinology and Metabolism. Arch Endocrinol Metab 2018; 62: 236-63.
13. Valdes-Socin H, Potorac I, Janin N, Bourguignon JP, Beckers A. Macroprolactinome pédiatrique sporadique associé à une mutation germinale AIP R304Q : Rémission quatre ans après l’interruption d’un traitement par Cabergoline. En: http://hdl.handle.net/2268/174088; consultado septiembre 2019.
14. Leontiou CA, Gueorguiev M, van der Spuy J, et al. The role of the aryl hydrocarbon receptor-interacting protein gene in familial and sporadic pituitary adenomas. J Clin Endocrinol Metab 2008; 93: 2390-401.
15. Vroonen L, Daly AF, Beckers A. Epidemiology and management challenges in prolactinomas. Neuroendocrinology 2019; 109: 20-7.