
LUCÍA VARELA 1, MARCELO SERRA 2, JUAN IGNACIO ROJAS 3, LILIANA PATRUCCO 1, AGUSTÍN PAPPOLLA 1
1 Servicio de Neurología, 2 Servicio de Clínica Médica, Hospital Italiano de Buenos Aires, 3 Centro de Esclerosis Múltiple de Buenos Aires, Servicio de Neurología, Hospital Universitario CEMIC, Buenos Aires, Argentina
Resumen Las enfermedades del espectro neuromielitis óptica son trastornos inflamatorios del sistema nervioso central caracterizados por una grave desmielinización y daño axonal inmunomediado que afecta principalmente a los nervios ópticos y médula espinal. Suelen presentars e en edades tempranas, aunque existen algunas comunicaciones en la literatura de pacientes con presentaciones tardías. Presentamos el caso de una mujer de 78 años que consultó por un cuadro de paraparesia grave, trastornos sensitivos y retención urinaria. Se realizó una resonancia magnética de columna cervicodorsal que evidenció una lesión medular longitudinal extensa. Se descartaron otras causas secundarias, basadas en la clínica y en resultados de laboratorio. El dosaje de anticuerpos anti-acuaporina 4 resultó positivo. Se indicó tratamiento con glucocorticoides a altas dosis y plasmaféresis, y mantenimiento con rituximab, obteniendo escasa respuesta clínica. En pacientes con lesiones medulares extensas se deben contemplar múltiples diagnósticos diferenciales según la presentación clínica, hallazgos mediante estudios por imágenes y epidemiología. Asimismo, debe incluir la búsqueda de anticuerpos anti-acuaporina 4 y contra la glicoproteína de la mielina del oligodendrocito, ya que el pronóstico funcional de estos pacientes suele ser desfavorable debido al gran componente destructivo de las lesiones. En consecuencia, el tratamiento temprano es fundamental a fin de limitar el daño agudo y prevenir futuras recaídas, lo cual es especialmente importante en presentaciones tardías de esta entidad debido a la escasa reserva funcional y baja capacidad de remielinización.
Palabras clave: neuromielitis óptica, mielitis longitudinalmente extensa, mielitis transversa
Abstract Optic neuromyelitis spectrum diseases are inflammatory disorders of the central nervous system characterized by severe demyelination and immunomediated axonal damage that mainly affects the optic nerves and spinal cord. They usually appear at an early age, although there are some reports in the literature of patients with late presentations. We present the case of a 78-year-old woman who consulted for severe paraparesis, sensory disorders, and urinary retention. An MRI of the cervicodorsal spine was performed, showing extensive longitudinal spinal injury. Secondary causes based on clinical observations and laboratory studies were ruled out. The dosage of anti-aquaporin 4 antibodies was positive. Acute treatment with high-dose glucocorticoids and plasmapheresis was indicated, and maintenance with rituximab, obtaining little clinical response. In patients with extensive spinal injuries, multiple differential diagnoses should be considered according to the clinical presentation, findings through imaging studies and epidemiology. Likewise, it should include the search for anti-aquaporin 4 antibodies and against the oligodendrocyte myelin glycoprotein, since the functional prognosis of these patients is usually unfavourable due to the large destructive component of the lesions. Consequently, early treatment is essential in order to limit acute damage and prevent future relapses, which is especially important in late presentations of this entity due to the low functional reserve and low remyelination capacity.
Key words: optic neuromyelitis spectrum disorders, transverse myelitis, longitudinally extensive transverse myelitis
Dirección postal: Lucía Varela, Servicio de Neurología, Hospital Italiano de Buenos Aires, Tte. Gral. Juan D. Perón 4190, 1199 Buenos Aires, Argentina
e-mail: lucia.varela@hospitalitaliano.org.ar
El espectro neuromielitis óptica (NMO) es una entidad descrita por primera vez en el año 2007 que incluye una serie de síndromes clínicos caracterizados por lesiones inflamatorias en diferentes topografías del sistema nervioso central, particularmente en el nervio óptico y médula espinal. Allí se otorga especial importancia a la presencia de anticuerpos anti-acuaporina 4 (AQP4) como responsables patogénicos directos de una proporción importante de casos 1. Sin embargo, en la última década se identificaron de forma creciente pacientes seronegativos con lesiones desmielinizantes atribuibles al espectro NMO a nivel supratentorial, diencéfalo y del tronco encefálico, ampliándose su rango de presentación clínica 2, 3.
Al igual que otros trastornos autoinmunes del sistema nervioso central, suele presentarse en edades tempranas, mediante fenómenos altamente agresivos que ocasionan una morbilidad significativa por la acumulación de recaídas clínicas. No obstante, su presentación puede producirse en sujetos mayores en una minoría de los casos, denominada NMO de presentación tardía. Presentamos un caso clínico de comienzo tardío de esta entidad y describimos su enfoque clínico y terapéutico.
Caso clínico
Mujer de 78 años con antecedentes de hipertensión arterial y dislipemia que cursó internación reciente por neumonía aguda de la comunidad. En dicho contexto se halló un nódulo pulmonar solitario (NPS) que continuó en estudio al alta tras resolver su cuadro infeccioso. Dos semanas después de su externación consultó por parestesias braquiocrurales y paraparesia de 4 días de evolución. Al examen físico se constató paraparesia grave, nivel sensitivo en segmento T3, retención urinaria e hipotonía del esfínter anal externo. Los estudios básicos de laboratorio (hemograma, función renal, ionograma, hepatograma y estado ácido base) al ingreso fueron normales. Se realizó una resonancia magnética (RMN) de cerebro y columna que evidenció una lesión centromedular longitudinalmente extensa entre los niveles C6 y T12, con refuerzo nodular heterogéneo tras la administración de contraste (Fig. 1). Con el propósito de descartar causas secundarias de mielopatía longitudinalmente extensa (MLE) se solicitó el estudio de anticuerpos anti cardiolipinas, coagulante lúpico, beta-2 glicoproteína, músculo liso, células parietales, factor intrínseco, nucleares, Ro y La, serología para HIV y sífilis, vitamina B12 y cobre sérico, que resultaron normales. Por su antecedente infeccioso se solicitaron anticuerpos IgM para Chlamydia y Mycoplasma pneumoniae, también negativos. Se realizó estudio del líquido cefalorraquídeo (LCR) en donde se destacó una leucorraquia de 604/mm3 (80% neutrófilos), hiperproteinorraquia de 327 mg/dl, glucorraquia de 44 mg/dl (glucemia sérica de 120 mg/dl) y aumento del lactato. En virtud de estos hallazgos, se solicitaron estudios moleculares por PCR para Herpes 1 y 2, 6, 8, varicela zoster, tuberculosis, citomegalovirus, Epstein-Barr y enterovirus, sin particularidades. El cultivo y análisis micológico tampoco mostraron alteraciones.
La determinación de bandas oligoclonales (BOC) reveló distribución policlonal en suero y LCR (tipo 1). Ante la sospecha de MLE paraneoplásica se decidió profundizar el estudio del NPS mediante PET-TC, que resultó discretamente hipermetabólico en dicha topografía. Se decidió realizar una punción biopsia del mismo, cuyo análisis histopatológico resultó inespecífico.
Se completó su estudio con anticuerpos onconeuronales en suero y LCR, también negativos. Por último, se decidió solicitar determinación de anticuerpos IgG anti glicoproteína de mielina del oligodendrocito (MOG) y acuaporina-4 por células transfectadas, siendo esta última positiva.
La paciente realizó tratamiento con 5 pulsos de 1 gramo de metilprednisolona y 6 sesiones de plasmaféresis. Luego continuó con descenso corticoide progresivo durante 4 semanas, sin obtener respuesta neurológica significativa. Se decidió realizar seguimiento clínico del nódulo pulmonar solitario e iniciar tratamiento con rituximab para disminuir el riesgo de nuevas recaídas clínicas.
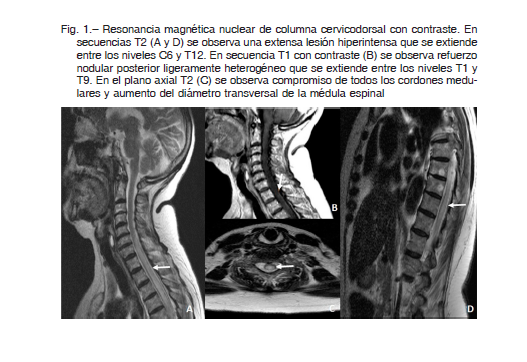
Discusión
Las enfermedades del espectro NMO son trastornos autoinmunes caracterizados por recaídas inflamatorias
agresivas en diferentes topografías del sistema nervioso central. Los eventos clínicos característicos son la neuritis óptica y mielitis inflamatoria aguda (frecuentemente MLE). Sin embargo, pueden presentarse además lesiones en el área postrema, tronco encefálico, diencéfalo o parénquima cerebral 2. En la actualidad, el diagnóstico se basa en los criterios revisados del año 2015, que requieren al menos un evento clínico y la detección de anticuerpos AQP4. En pacientes seronegativos, se necesitan al menos dos eventos clínicos con diseminación en espacio, uno de los cuales debe ser característico, y lesiones sugerentes en RMN2. Una proporción menor de ellos puede mostrar positividad para anticuerpos anti-MOG.
La enfermedad es más prevalente en el sexo femenino (9-10:1) 3 y la edad más frecuente de presentación oscila entre los 20 y 45 años. Sin embargo, existen comunicaciones crecientes en la literatura sobre casos de presentación tardía y muy tardía, donde las edades de presentación superan los 50 y 75 años, respectivamente. En consonancia con nuestro caso, el pronóstico funcional de estos pacientes suele ser desfavorable, debido a la escasa reserva funcional previa y baja capacidad de remielinización 4-7. Desde el punto de vista geográfico, es más frecuente en poblaciones de Asia, África y Latinoamérica que en la población de origen caucásico. Existen pocos datos sobre su comportamiento en nuestro medio 8, aunque es probable que en la Argentina sea menos frecuente que en otros países de la región debido a la alta tasa de ascendencia europea 9.
El diagnóstico de pacientes con MLE debe contemplar la búsqueda de anticuerpos anti-AQP4 y MOG así como otros diagnósticos diferenciales según la presentación clínica, hallazgos mediante estudios por imágenes y epidemiología 10, 11. Entre los más frecuentes, se encuentran las enfermedades inflamatorias autoinmunes como esclerosis múltiple, encefalomielitis diseminada aguda, lupus eritematoso sistémico, sarcoidosis y síndrome de Sjögren. Finalmente existen otras etiologías infecciosas, neoplásicas, vasculares y metabólicas que pueden causar cuadros clínicos e imágenes similares a una MLE, cuyo diagnóstico de certeza es mandatorio ya que el tratamiento varía según la etiología (Tabla 1). Particularmente en nuestro medio, sugerimos considerar micosis sistémicas endémicas, HTLV-1, sífilis y tuberculosis. En caso de que las determinaciones precedentes resulten negativas o alta sospecha de enfermedad oncológica, se deberá evaluar la realización de PET-TC y eventualmente anticuerpos antineuronales, debido a que ciertas enfermedades paraneoplásicas son también capaces de causar MLE 11.
El estudio del LCR puede resultar orientador en el diagnóstico de MLE debido a trastornos inflamatorios.
En el espectro NMO puede existir pleocitosis mayor a 50 leucocitos/ml en un 35% de los individuos. Además, el hallazgo de neutrófilos o eosinófilos mayor a 5 células/ml ocurre entre un 10-44% de los casos. La presencia de BOC en LCR puede detectarse en pacientes con espectro NMO hasta en un 20-30%, mientras que en pacientes con EM el número asciende al 80% 2,11. Por último, la proteína ácida fibrilar glial (GFAP) suele encontrarse elevada en LCR en pacientes con espectro NMO, pero solo durante algunos días o semanas posteriores al ataque.
El tratamiento agudo de la MLE del espectro NMO suele iniciarse con corticoides endovenosos a altas dosis con eventual plasmaféresis en simultáneo o de forma subsiguiente según la respuesta clínica. Una vez concluida esta fase, es mandatorio controlar de forma sostenida el componente inmunomediado. En este sentido, los tratamientos con mayor eficacia son rituximab, azatioprina y micofenolato 12. Si bien no existen estudios a la fecha que evalúen la seguridad y eficacia de estos tratamientos en presentaciones tardías de la enfermedad, la utilización de los mismos se indica igualmente en función de la extrapolación de datos conocidos en relación a estudio retrospectivos.
El espectro NMO debe ser considerado siempre en un paciente con mielopatía aguda, especialmente MLE. Si bien esta entidad no es frecuente en pacientes añosos y es mandatorio descartar en primera instancia otras etiologías más prevalentes, debe ser tenida en cuenta entre las posibilidades diagnósticas, con búsqueda de anticuerpos anti AQP4 y MOG. En este sentido, el correcto enfoque de estos pacientes permitirá iniciar tratamiento inmunomodulador destinado a limitar la aparición de futuras recaídas, cuyo pronóstico es particularmente desfavorable en edades avanzadas.

Conflicto de intereses: Ninguno para declarar
Bibliografía
1. Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol 2007; 6: 805-15.
2. Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015; 85: 177-89.
3. Pedraza M, Pérez MA, Navarrete L, et al. Trastorno del espectro de la neuromielitis óptica. Rev Salud Bosque 2019; 9: 99-105.
4. Seok JM, Cho HJ, Ahn SW, et al. Clinical characteristics of late-onset neuromyelitis optica spectrum disorder: A multicenter retrospective study in Korea. Mult Scler 2017; 23: 1748-56.
5. Mao Z, Yin J, Zhong X, et al. Late-onset neuromyelitis optica spectrum disorder in AQP4-seropositive patients in a Chinese population. BMC Neurol 2015; 15: 160.
6. Lefaucheur R, Bourre B, Ahtoy P, et al. Neuromyelitis optica with very late onset. J Am Geriatr Soc 2011; 59: 1138-40.
7. Krumbholz M, Hofstadt-van Oy U, Angstwurm K, et al. Very late-onset neuromyelitis optica spectrum disorder beyond the age of 75. J Neurol 2015; 262: 1379-84.
8. Alvarenga MP, Schimidt S, Alvarenga, RP. Epidemiology of neuromyelitis optica in Latin America. Mult Scler J Exp Transl Clin 2017; 3: 2055217317730098.
9. Zarei S, Eggert J, Franqui-Dominguez L, et al. Comprehensive review of neuromyelitis optica and clinical characteristics of neuromyelitis optica patients in Puerto Rico. Surg Neurol Int 2018; 9: 242.
10. Cho TA, Bhattacharyya S. Approach to myelopathy. Continuum (Minneap Minn) 2018; 24: 386-406.
11. Wingerchuk DM. Immune-mediated myelopathies. Continuum (Minneap Minn) 2018; 24: 497-522.
12. Flanagan EP. Neuromyelitis Optica Spectrum Disorder and Other Non-Multiple Sclerosis Central Nervous System Inflammatory Diseases. Continuum (Minneap Minn) 2019; 25:815-44.
– – – –
…creativity in science, as in the arts, cannot be organised. It arises spontaneously from individual talent. Well-run laboratories can foster it, but hierarchical organization, inflexible, bureaucratic rules, and mountains of futile paperwork can kill it. Discoveries cannot be planned; they pop up, like Puck, in unexpected corners.
…creatividad en la ciencia, como en las artes, no puede ser organizada. Surge espontáneamente del talento individual. Laboratorios bien llevados pueden promoverla, pero organizaciones arcaicas, reglas burocráticas, y montañas de expedientes pueden aniquilarla. Los descubrimientos no pueden ser planeados, suelen surgir, como Puck, en rincones inesperados.
Max Perutz
I wish I´d made you angry earlier. Cold Spring Harbor: Cold Spring Harbor Laboratory Press, 1998, p IX