
PEDRO J. QUIROGA-PADILLA 1, PAULA V. GAETE 1, CARLOS O. MENDIVIL 1, 2
1 Universidad de los Andes, Facultad de Medicina, 2 Sección de Endocrinología, Fundación Santa Fe de Bogotá, Bogotá, Colombia
Resumen La quilomicronemia familiar es una condición en que una mutación genética altera la capacidad de metabolizar los triglicéridos que viajan en las lipoproteínas, causando elevación extrema de triglicéridos plasmáticos y complicaciones asociadas. La complicación más frecuente es la pancreatitis, que puede llevar a falla multiorgánica o insuficiencia pancreática. La quilomicronemia familiar también afecta la calidad de vida, las relaciones sociales y el desarrollo profesional. El gen más frecuentemente afectado en la quilomicronemia familiar es el de lipoproteína lipasa-1 (LPL), enzima que hidroliza triglicéridos circulantes para su captación tisular. Mutaciones en genes (como APOC2, APOAV, LMF-1, GPIHBP-1) que codifican para proteínas que regulan la maduración, transporte o polimerización de lipoproteína lipasa-1, también pueden estar involucradas. Sin embargo, en cerca del 30% de los pacientes no se encuentra la variante causal. La quilomicronemia familiar debe sospecharse en casos de hipertrigliceridemia extrema, resistente al tratamiento convencional, o que se acompaña de xantomas eruptivos, lipemia retinalis o dolor abdominal. La disponibilidad de escalas de riesgo y pruebas genéticas deben promover la detección oportuna. La nutrición se basa en una dieta muy baja en grasa con adecuada suplencia de vitaminas liposolubles y ácidos grasos esenciales, además de evitar el consumo de alcohol. Si bien el tratamiento farmacológico incluye fibratos y ácidos grasos omega 3, el enfoque actual privilegia agentes biotecnológicos dirigidos a los defectos moleculares propios de la enfermedad. Ello incluye un oligonucleótido antisentido dirigido contra apoC-III (volanesorsen), un anticuerpo monoclonal contra la proteína similar a angiopoietina tipo 3 (evinacumab), y otros compuestos en desarrollo.
Palabras clave: quilomicronemia, triglicéridos, hipertrigliceridemia, dislipidemias, apoC-III, volanesorsen
Abstract Familial chylomicronemia is a disease in which a genetic mutation affects the ability of the organism to metabolize triglycerides bound to lipoproteins, causing extremely high plasma triglycerides and associated consequences. The most frequent complication is acute pancreatitis, which may lead to multiorganic failure or pancreatic insufficiency. Familial chylomicronemia also exerts a profound negative impact on quality of life, social relationships and professional development. The gene most frequently affected is lipoprotein lipase-1 gene (LPL), the enzyme in charge of hydrolyzing circulating triglycerides for tissue uptake. Mutations in other genes regulating maturation, transport or polymerization (eg. APOC2, APOAV, LMF-1, GPIHBP-1) of lipoprotein lipase-1, may also be involved. However, in about 30% of patients the causal variant is not identified. Familial chylomicronemia should be suspected in patients with severe hypertriglyceridemia with poor response to conventional treatment, or accompanied by eruptive xanthomas, lipemia retinalis or abdominal pain. The availability of risk scores and genetic tests should facilitate its opportune detection and management. Nutritional therapy is based on a very-low-fat diet with adequate supply of lipid-soluble vitamins and essential fatty acids, plus avoidance of alcohol consumption. Current pharmacological treatment may include fibrates and omega-3 fatty acids but prioritizes biotechnological agents targeting the molecular disturbances of the disease. These include an antisense oligonucleotide against apoC-III (volanesorsen), a monoclonal antibody against angiopoietin-like protein-3 (evinacumab), and other agents currently in development.
Key words: chylomicronemia, triglycerides, hypertriglyceridemia, dyslipidemias, apoC-III, volanesorsen
Dirección postal: Carlos O. Mendivil, Facultad de Medicina, Universidad de los Andes, Carrera 7 N° 116-05, Of. 413, Bogotá, Colombia
e-mail: carlosolimpo@gmail.com
La quilomicronemia familiar es una enfermedad poco frecuente, pero con serias implicaciones y consecuencias, que se pueden prevenir a través de un reconocimiento temprano y una atención oportuna. Aproximadamente la mitad de los pacientes con quilomicronemia familiar sienten que sus médicos no entienden qué es la enfermedad, e incluso, el 30% considera que el tratamiento dado por su médico empeoró los síntomas 1. En esta revisión, buscamos realizar una puesta al día de la etiología, así como del diagnóstico y tratamiento actuales de esta condición.
Metabolismo normal de triglicéridos
Los lípidos son compuestos insolubles en agua que tienen diferentes funciones en el cuerpo humano. Existen varios tipos: los triglicéridos (TG), los fosfolípidos, los esfingolípidos y los esteroles (como el colesterol). Los TG son la forma química en la que los ácidos grasos son transportados en circulación, están formados por un esqueleto de glicerol y tres ácidos grasos que se adicionan a cada uno de sus carbonos. Estos ácidos grasos luego pueden ser utilizados como sustrato energético, como molécula de señalización celular o como componente estructural de otros lípidos (fosfolípidos, glicolípidos o ésteres de colesterol) 2.
Debido a que los TG son insolubles en agua, necesitan asociarse a proteínas para poder ser transportados a través del torrente sanguíneo. A esta asociación entre lípidos y proteínas se denomina lipoproteína. Las lipoproteínas son partículas complejas que en su interior están formadas por un centro o core que contiene ésteres de colesterol (una molécula de colesterol con un ácido graso) y TG, y una superficie que contiene colesterol libre, fosfolípidos y apoproteínas (las proteínas que forman parte de las lipoproteínas). Las apoproteínas actúan como ligandos de receptores y como activadores o inhibidores de enzimas involucradas en el metabolismo de lípidos. Las lipoproteínas se clasifican según su tamaño, densidad y composición en diferentes tipos (Tabla 1). Las lipoproteínas ricas en TG son los quilomicrones (QM) y las lipoproteínas de muy baja densidad (VLDL), las primeras transportan a los TG de la dieta, mientras que las segundas a los TG sintetizados a nivel hepático 3.
Los ácidos grasos se clasifican según su longitud en ácidos grasos de cadena corta (menos de 6 carbonos), media (6 a 12 carbonos) y larga (más de 12 carbonos).
Si bien la digestión y absorción de todos los TG dietarios es similar, existen diferencias según el tipo de ácido graso que conforme dichos TG. Los TG conformados por ácidos grasos de cadena corta y media son hidrolizados a glicerol y ácidos grasos libres, son absorbidos por los enterocitos y luego cada ácido graso se une a la albúmina y es transportado al hígado a través de la vena porta 4.
Por otra parte, los TG con ácidos grasos de cadena larga son digeridos a 2-monoacilglicerol y ácidos grasos libres, que ingresan al enterocito, en donde se reensamblan y se empacan en los QM junto con la apoproteína B48. Los QM acceden a los conductos linfáticos llegando hasta el conducto torácico y luego al sistema venoso. Debido a que los QM están formados por los ácidos grasos de la dieta, siempre hay quilomicronemia transitoria después de ingerir grasas. Sin embargo, en condiciones normales no debería existir quilomicronemia en el estado de ayuno 5.
Las VLDL son lipoproteínas ricas en TG, sintetizadas y secretadas por el hígado bajo el estímulo de la insulina.
Las VLDL son formadas con apolipoproteína B-100 (apoB100) y TG endógenos, producidos por el hígado a partir de los carbohidratos de la dieta. Además de una ingesta elevada de carbohidratos, el consumo excesivo de alcohol y altos niveles de estrógenos también estimulan de forma independiente la síntesis de VLDL, por lo que pueden causar hipertrigliceridemia endógena6, completamente diferente a la quilomicronemia familiar de la que se ocupa este artículo (Fig. 1).
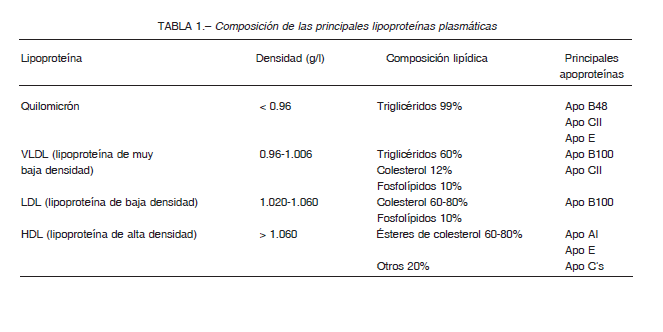
Después de ser sintetizadas, tanto VLDL como QM son metabolizados de la misma forma. Ambas interaccionan con las HDL y sirven como sustrato de una proteína que media el intercambio de lípidos: la proteína de transferencia de ésteres de colesterol (PTEC). La función de esta proteína es llevar ésteres de colesterol de las HDL a los QM y VLDL, y simultáneamente llevar TG de las VLDL y QM a las HDL. En esta interacción, HDL también le cede a QM y VLDL dos apoproteínas muy importantes para su metabolismo: apoproteína E (apoE) y apoproteína C-II (apoC-II). ApoE se requiere para la captación hepática de lipoproteínas, mientras que apoC-II es cofactor de la lipoprotein-lipasa-1 (LPL-1), la enzima presente en la superficie de las células endoteliales que procesa los TG circulantes y permite que los ácidos grasos sean entregados a los tejidos periféricos 2 (Fig. 2).
Causas moleculares de la quilomicronemia familiar
La causa más frecuente de quilomicronemia familiar son las mutaciones en el gen de la LPL-1. Este gen se encuentra en el brazo pequeño del cromosoma 22 y está conformado por 10 exones. Se han encontrado diferentes tipos de mutaciones que pueden llevar a una disminución en la función de esta enzima, un aumento en el riesgo de enfermedad cardiovascular, pancreatitis y un perfil lipídico desfavorable, consistente en hipertrigliceridemia y concentraciones bajas de colesterol de HDL. La deficiencia genética de LPL-1 causa quilomicronemia familiar con un patrón de herencia autosómico recesivo 2.
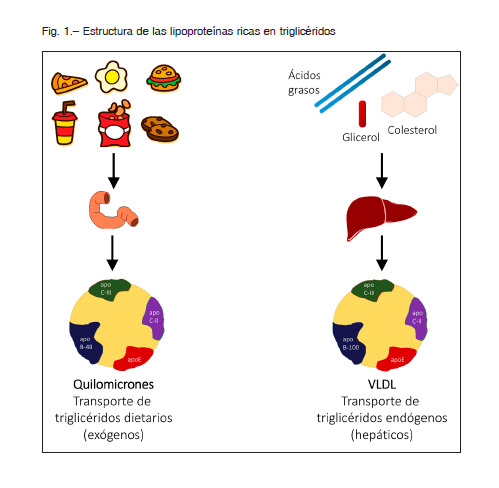
La actividad de la LPL-1 está determinada por la acción de distintas proteínas. Después de ser producida en células musculares o adipocitos, la LPL-1 debe glucosilarse y dimerizarse en un proceso liderado por el factor de maduración de lipasa-1 (LMF1 – por su nombre en inglés). Otros factores que influencian la actividad de LPL-1 son:
– Las proteínas similares a la angiopoietina -3, -4 y -8 (ANGPTL-3, -4 y -8 – por sus nombres en inglés), en general son inhibidores endógenos de LPL-1 7.
– La apoproteína C-III (apoC-III), parece ser también un inhibidor de LPL-1 y especialmente un antagonista de apoE 8.
– La apoproteina A-V (apoA-V), necesaria para estabilizar los complejos de LPL-1, que in vivo funciona como un multímero 9.
– La proteína de unión a la HDL anclada a glucosilfosfatidilinositol tipo 1 (GPIHBP-1 por su nombre en inglés), que transloca la LPL-1 a la superficie endotelial y simultáneamente ancla los QM al endotelio, permitiendo así la interacción de LPL-1 con los QM 10 (Fig. 2).
Las proteínas similares a angiopoietina inhiben la acción de LPL-1 al promover la disociación de dímeros de LPL-1; la ANGPTL3 es la inhibidora más eficaz, mientras que ANGPTL4 es la más potente 11. Estudios en humanos han identificado que mutaciones de pérdida de función del gen ANGPTL3 se asocian a bajos niveles plasmáticos de TG, mientras que concentraciones elevadas de la proteína ANGPTL3 se relacionan con hipertrigliceridemia e hipercolesterolemia 12, 13. En línea con lo anterior, niveles plasmáticos elevados de apoC-III 14 y disminuidos de apoA-V, así como la presencia de mutaciones que resulten en pérdida de su función, se relacionan con hipertrigliceridemia 15, 16. La importancia de la función de GPIHBP-1 se ha evidenciado en estudios con ratones GPIHBP-1 -/-, que presentan hipertrigliceridemia grave 17.
Definición de hipertrigliceridemia y quilomicronemia
La hipertrigliceridemia es un hallazgo frecuente en la práctica clínica, pero su definición a partir de un punto de corte único es motivo de debate. Sin embargo, cuando los niveles de TG superan los 500 mg/dl, lo más frecuente es que exista algún grado de presencia de QM en la sangre 18. La quilomicronemia se define como la elevación persistente (al menos tres mediciones consecutivas) de QM en plasma después de un periodo de ayuno de 12 a 14 horas 19,20. La vida media normal de un QM en sangre es de tan solo 5 minutos 21, por lo que no deberían encontrarse QM en sangre tras un periodo de ayuno prolongado.
Etiología de la hipertrigliceridemia y la quilomicronemia
La etiología de la hipertrigliceridemia es un área de investigación con un avance vertiginoso en los años recientes.
La hipertrigliceridemia y la quilomicronemia tienen una etiología compleja donde median factores genéticos (Tablas 2 y 3) 19 y ambientales 22. Las causas más frecuentes de hipertrigliceridemia o quilomicronemia secundarias son la diabetes tipo 2, el consumo excesivo de alcohol, la terapia hormonal con estrógenos, el embarazo y ciertos medicamentos como beta-bloqueadores no específicos, isotretinoína, inhibidores de la proteasa de VIH, ciclosporina y algunos antipsicóticos atípicos 20, 22.
Los valores plasmáticos de TG son un espectro continuo, se estima que aproximadamente el 30% de las personas de la población general tiene hipertrigliceridemia 23.
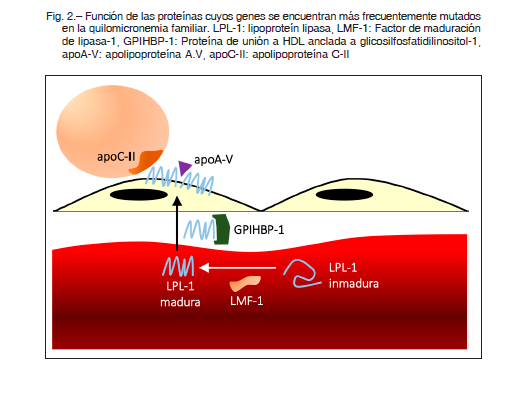
Se han informado 32 genes cuyas variantes están relacionadas con el nivel de TG en plasma 24. Dichas variantes se clasifican en comunes y raras, y las variantes raras tienen un efecto mucho más marcado sobre el nivel de TG en comparación a las variantes comunes. El efecto acumulativo de variantes comunes y raras, junto con factores ambientales, da como resultado la gran variación observada en los valores de TG22 en la población general. Las variantes con un mayor efecto cuantitativo sobre los triglicéridos se resumen en la Tabla 3. Los pacientes con TG elevados a causa de variantes comunes tienen hipertrigliceridemia poligénica, mientras que personas con una variante rara en genotipo homocigoto o heterocigoto compuesto tienen quilomicronemia familiar 22.
En la población general, la etiología más frecuente de hipertrigliceridemia es la poligénica 25.
El síndrome de quilomicronemia familiar resulta de mutaciones en uno o más genes involucrados en la lipólisis o la remoción de los quilomicrones circulantes 19. Los seis genes afectados con mayor frecuencia son LPL, APOC2, APOA5, GPIHBP-1 y LMF1 (Tabla 2), aunque aproximadamente en el 30% de los pacientes no se logra identificar una variante causal específica 18, 26. La prevalencia de quilomicronemia familiar en diferentes poblaciones se ha estimado entre 1/6000 para centros de alta complejidad hasta 1/300 000 en la población general 27, en tanto que la prevalencia de hipertrigliceridemia poligénica grave se ha estimado en 1/600 personas 19.
Consecuencias clínicas de la quilomicronemia familiar
La relevancia clínica de la quilomicronemia familiar radica en sus complicaciones. La complicación más grave de la quilomicronemia es la pancreatitis aguda, ya que genera desenlaces fatales hasta en un 30% de los casos y una alta carga de morbilidad dada por falla multiorgánica, necrosis o insuficiencia pancreática, absceso o pseudoquiste pancreático y pancreatitis crónica 18, 19. A pesar de ser la mayor consecuencia, la pancreatitis aguda es prevenible.
El riesgo de pancreatitis aguda aumenta drásticamente si los TG plasmáticos superan 885 mg/dl 19: cuatro por ciento por cada 100 mg/dl de aumento en los TG 20. En cuanto al riesgo cardiovascular, en los pacientes con quilomicronemia familiar la aterosclerosis prematura es rara, probablemente porque los QM son demasiado grandes para penetrar al endotelio 18, 21, 22, 28, 29. No obstante, en la población general los niveles anormalmente elevados de TG sí se asocian con un mayor riesgo cardiovascular, sobre todo debido al riesgo conferido por las lipoproteínas remanentes (VLDL y quilomicrones parcialmente metabolizados) 30.
La quilomicronemia familiar también tiene consecuencias sociales y un efecto negativo en calidad de vida. En un estudio que involucró 10 países y 166 participantes, se encontró que 43% de los pacientes con quilomicronemia familiar sienten que la enfermedad interfiere con su habilidad para socializar, 40% de ellos se encontraban desempleados al momento de la encuesta, de los cuales 65% atribuyeron su condición de desempleo a la quilomicronemia 1.
Diagnóstico de la quilomicronemia familiar
Los motivos de consulta más comunes de pacientes con quilomicronemia son el hallazgo de hipertrigliceridemia grave y/o un episodio de pancreatitis aguda 28. El diagnóstico de quilomicronemia familiar es un verdadero reto: en un estudio de pacientes diagnosticados con esta enfermedad, cada uno consultó en promedio a 5 especialistas distintos (rango: 1-30) antes de obtener el diagnóstico definitivo 31. Incluso algunos pacientes cuya presentación inicial fue un abdomen agudo, han sido sometidos innecesariamente a cirugías abdominales 32, 33.
Se debe sospechar quilomicronemia familiar en pacientes con importante hipertrigliceridemia, especialmente si no es secundaria a una causa identificable o si se acompaña de pancreatitis aguda 34.

Tanto la hipertrigliceridemia poligénica como la quilomicronemia familiar pueden presentarse con xantomas eruptivos, lipemia retinalis, hepatoesplenomegalia, dolor abdominal, náusea y vómito, y pancreatitis aguda 18. Otras manifestaciones menos comunes incluyen palidez mucocutánea, anemia e irritabilidad 18. Los xantomas eruptivos son pápulas amarillentas de macrófagos cargados de lípidos (células espumosas) rodeadas por halos eritematosos que aparecen usualmente en las áreas extensoras de las extremidades, glúteos y hombros. La lipemia retinalis se caracteriza por la apariencia blanquecina/rosada de los vasos retinianos secundaria al alto contenido de QM, sin comprometer la agudeza visual 18. De igual importancia son los síntomas cognitivos y emocionales, así como las implicaciones sociales: la quilomicronemia familiar se asocia con dificultad para concentrarse, ansiedad constante por la incertidumbre de tener un nuevo episodio de pancreatitis y una alta percepción de interferencia con las relaciones sociales 1, 31.
Hasta un 85% de los pacientes con hipertrigliceridemia grave de cualquier etiología desarrollarán pancreatitis aguda en algún momento de su evolución 19, 25, 32. Es importante tener en cuenta la elevación marcada de TG, ya que puede afectar los equipos de medición e inducir a subestimación de los niveles de amilasa, electrolitos y hemoglobina 21. Por lo anterior, niveles normales de amilasa no excluyen el diagnóstico de pancreatitis aguda en este contexto. El mecanismo por el cual la hipertrigliceridemia causa pancreatitis aguda no está completamente elucidado, sin embargo, se considera que la acumulación de QM en los capilares pancreáticos facilita la lipólisis de TG por la lipasa pancreática. Los ácidos grasos liberados tienen efectos citotóxicos directos sobre el parénquima o llevan a activación del tripsinógeno y subsecuente autodigestión del tejido pancreático 18, 19, 34. La diferencia clínica más importante entre hipertrigliceridemia poligénica y quilomicronemia familiar es la edad de presentación, la primera aparece usualmente en la edad adulta, mientras que la segunda lo hace más frecuentemente en la infancia o en la adolescencia 1, 28, 33.
Los TG plasmáticos no sirven para diferenciar hipertrigliceridemia poligénica de la quilomicronemia familiar, pueden encontrarse extremadamente altos en cualquiera de las dos condiciones. Un hallazgo de laboratorio que puede orientar es que los pacientes con hipertrigliceridemia poligénica pueden tener niveles más elevados de colesterol total respecto a los pacientes con quilomicronemia familiar, ya que las VLDL contienen algo de colesterol, mientras que los QM no 28. Teniendo en cuenta que apoB-100 es una proteína estructural de las VLDL y LDL, pero no de los QM (que tienen apoB-48), se ha propuesto que la concentración de apoB-100 en sangre puede diferenciar entre quilomicronemia familiar y otras hipertrigliceridemias graves. Un paciente con TG extremadamente altos y niveles de apoB-100 normales o solo ligeramente elevados, debe despertar la sospecha de quilomicronemia familiar 20, 32, 34-36. En la práctica clínica también se emplea como subrogado de la concentración de QM una razón entre TG y colesterol total mayor a 5.
Antes del desarrollo de la secuenciación de ADN por la técnica de Sanger, la quilomicronemia familiar era diagnosticada por la ausencia de actividad de LPL-1 en plasma recolectado después de la administración intravenosa de heparina 37. Hoy en día la determinación de la actividad de LPL-1 se usa poco, y el estándar de oro para diagnosticar quilomicronemia familiar es el análisis genético 34.
En general se usan dos técnicas de secuenciación para detectar variantes en los genes involucrados: secuenciación Sanger o secuenciación de nueva generación (NGS por sus siglas en inglés) 38. En ambos casos se necesita una muestra de sangre, de la cual se aíslan los leucocitos para extraer y purificar su ADN genómico 39. Mientras que la secuenciación por Sanger se dirige a un único gen por ensayo, la NGS puede analizar varios genes o todo el genoma en un solo ensayo 38, 39. Como se mencionó antes, variantes en 5 genes causan la quilomicronemia familiar (en cada gen se han descrito de 10 a 156 variantes) 40.
Recientemente se desarrolló la prueba LipidSeq®, una prueba basada en NGS que permite secuenciar en la misma muestra 73 genes y 178 polimorfismos de un único nucleótido relacionados con dislipidemias. LipidSeq® disminuye los costos de secuenciación, identifica todas las variantes relacionadas a la actualidad con los TG y otras dislipidemias, reduce el tiempo de resultados a menos de un día y, en especial, incluye un amplio número de variantes para diagnosticar hipertrigliceridemia poligénica o quilomicronemia familiar 38.
Determinar con precisión el diagnóstico de quilomicronemia familiar es un reto en el contexto del sistema de salud. Solo el 6% de 166 pacientes con quilomicronemia familiar contactados por e-mail informaron tener diagnóstico genéticamente confirmado 1. Por esta razón, se desarrolló un puntaje basado en características clínicas y resultados paraclínicos para el diagnóstico de quilomicronemia familiar llamado el FCS score (de familial chylomicronemia syndrome score) (Tabla 4) 20. El FCS score incluye los valores históricos de TG, presencia o ausencia de factores que eleven secundariamente los TG, historia de pancreatitis aguda o dolor abdominal recurrente sin causa clara, historia familiar de hiperlipidemia familiar combinada, respuesta a tratamiento farmacológico para hipertrigliceridemia y edad al inicio de los síntomas. El FCS score tiene una sensibilidad de 87.5% y una especificidad entre 66.6 y 87.5%, para diferenciar entre hipertrigliceridemia poligénica y quilomicronemia familiar genéticamente diagnosticada 20.
Tratamiento no farmacológico
El tratamiento no farmacológico para la quilomicronemia familiar se basa en nutrición y actividad física, siendo el primer elemento mucho más importante. En cuanto a la actividad física, se aconseja seguir las recomendaciones generales de la Organización Mundial de la Salud 41.
Sin embargo, uno de los grandes desafíos del síndrome de quilomicronemia familiar es el tratamiento nutricional, que tiene como meta mantener niveles de ácidos grasos esenciales y vitaminas liposolubles adecuados, con una dieta muy baja en grasa y sin sobrecarga de carbohidratos. Una dieta muy baja en grasa es aquella en la que el consumo de grasa aporta menos del 15% de las calorías diarias totales 42, 43. Además de esto, es importante que como máximo el 60% de las calorías sean aportadas por los carbohidratos, ya que como se había explicado antes, las azúcares de la dieta pueden usarse como sustrato para formar ácidos grasos a nivel hepático, que luego pueden ser convertidos a triglicéridos y ser exportados en las VLDL, contribuyendo a aumentar aún más la trigliceridemia 6. Esta meta de carbohidratos se puede lograr consumiendo productos ricos en proteína y bajos en grasa (carnes magras de res, ave, cerdo o pescado sin piel, a excepción del salmón), lácteos bajos en grasa, huevos sin yema, alimentos con alta densidad calórica pero mínimo contenido de grasa como los vegetales, los granos integrales y las legumbres; alimentos altos en fibra, carbohidratos complejos más que carbohidratos simples, al mismo tiempo que se limita el consumo de frutas a entre 1 y 4 porciones al día, evitando las frutas más ricas en carbohidratos como uvas o banano y evitando los jugos de fruta y los alimentos con azúcar añadida 22, 43.

Dado que mantener un consumo de macronutrientes adecuado únicamente con estos alimentos puede ser complicado, se pueden usar suplementos de ácidos grasos de cadena media (ácido caprílico de 8 carbonos y ácido cáprico de 10 carbonos), ya que como se había explicado antes, estos ácidos grasos se transportan unidos a la albúmina por la vena porta y no a través de los QM. Se debe tener en cuenta que la tolerancia a estos ácidos grasos varía de persona a persona, por lo que se debe empezar con pequeñas cantidades e ir aumentando la ingesta 44.
El mantenimiento de la ingesta adecuada de ácidos grasos esenciales es especialmente importante para los niños, ya que una deficiencia de estos podría llevar a alteraciones del crecimiento y del desarrollo neurológico 43.
Los ácidos grasos esenciales son el ácido linoleico (omega-6) y el alfa-linolénico (omega-3), que cumplen una función en la formación de nuevas células, por lo que son indispensables para la cicatrización de heridas, el crecimiento de las uñas y el cabello y el mantenimiento de una piel sana. Se recomienda que la ingesta de ácidos grasos esenciales sea entre el 2 y el 4% de la ingesta calórica total, fuentes alimenticias importantes son las semillas de chía, la linaza, las nueces, la soya y los granos integrales, así como los aceites vegetales 45.
Debido a que las fuentes alimenticias son escasas, se pueden prescribir entre 3 y 4 gramos diarios de suplemento de ácidos grasos esenciales, sin embargo, es importante aclarar que este consumo se debe incluir en el cálculo de las calorías aportadas por la grasa del total de ingesta calórica diaria 46. Adicionalmente, se deben monitorizar constantemente los niveles séricos de vitaminas liposolubles (A, D, E y K) y prescribir suplementos nutricionales si es necesario, debido a que las personas con quilomicronemia familiar tienen más riesgo de desarrollar déficit. Por una parte, una dieta restrictiva en grasas limita el aporte dietario, y por otra al no poder metabolizar eficientemente los QM, puede no existir una apropiada entrega tisular de las vitaminas liposolubles 6.
Por último, también se recomienda evitar el consumo de alcohol, ya que este aumenta la síntesis de ácidos grasos, empeorando con ello la hipertrigliceridemia y promoviendo la acumulación de TG a nivel hepático 22. Es también importante mantener un adecuado consumo de líquidos para estar hidratado, ayudar a la digestión y contribuir a una función pancreática normal. El consumo de agua recomendado en adultos es de 35 ml/kg de peso 47.
Tratamiento farmacológico
Dado que en la mayoría de los pacientes con quilomicronemia familiar existe una alteración genética de alguna de las moléculas involucradas en el catabolismo de los triglicéridos circulantes, las terapias farmacológicas tradicionales para la hipertrigliceridemia (fibratos, ácidos grasos omega 3, niacina) tienen una eficacia bastante limitada y son incapaces de reducir los triglicéridos plasmáticos hasta niveles que no representen un riesgo inminente para la salud. Por esta razón, las terapias con eficacia en quilomicronemia familiar suelen pertenecer al grupo de los medicamentos biológicos/biotecnológicos.
Oligonucleótidos antisentido: Un nuevo grupo de medicamentos biotecnológicos
Algunos nuevos medicamentos desarrollados para el tratamiento de la quilomicronemia familiar son oligonucleótidos antisentido. Los oligonucleótidos antisentido son moléculas de ácido nucleico de cadena simple que se unen por complementariedad de bases al ARN mensajero en la célula e inducen su degradación o impiden su traducción. Al hacerlo, los oligonucleótidos antisentido reducen la síntesis de la proteína objetivo. Los oligonucleótidos antisentido suelen tener una longitud entre 12 y 20 nucleótidos y tienen una secuencia de nucleótidos complementaria a la de su ARN mensajero objetivo.
Para hacer que estas moléculas sean más estables y así tengan un efecto terapéutico más prolongado, es posible introducir diversas modificaciones químicas en su cadena invariante 48. Una vez internalizados a la célula, los oligonucleótidos antisentido se unen a regiones del ARN mensajero objetivo formando un heterodúplex ADN-ARN. Este complejo recluta a una enzima llamada RNasa H1, una endonucleasa que destruye específicamente la cadena de ARN de este heterodúplex 49. Los agentes de este tipo tienden a concentrarse en el hígado, que es justamente el órgano donde la mayoría de las apolipoproteínas son producidas, inducen la degradación de su ARN mensajero objetivo y luego son degradados por vías completamente independientes de la vía del citocromo P450, lo que en teoría les confiere un menor riesgo de interacciones farmacológicas 49. Los llamados oligonucleótidos de segunda generación, categoría a la cual pertenecen la mayoría de los agentes actualmente en desarrollo para el tratamiento de los lípidos, tienen en sus moléculas de ribosa un grupo metil o metoxietil (en vez de un -OH) en su carbono 2´.
Volanesorsen: oligonucleótido antisentido dirigido contra apoC-III
Volanesorsen (también llamado ISIS 304801), es un oligonucleótido antisentido de segunda generación, complementario al ARN mensajero de apoC-III. Al frenar la producción de apoC-III, este agente reduce la secreción de lipoproteínas ricas en triglicéridos y facilita la remoción de dichas lipoproteínas por el hígado, aún en ausencia de LPL-150. Un estudio de fase 2 evaluó el efecto de volanesorsen en pacientes con hipertrigliceridemia severa no tratada (TG plasmáticos entre 350 y 2000 mg/dl, 41 recibieron volanesorsen y 16 recibieron placebo), y en pacientes con una dosis estable de fibrato y TG plasmáticos entre 225 y 2000 mg/dl (20 recibieron volanesorsen y 8 recibieron placebo) 51. En este estudio, volanesorsen a dosis de 100-300 mg por semana durante 13 semanas indujo reducciones dosis-dependientes de apoC-III plasmática, lo cual se tradujo en una disminución de los TG de ayuno entre 31.3 y 70.9%. La respuesta al agente fue consistente a las diferentes dosis empleadas, en contraste con la alta variabilidad interindividual que se ha observado con otros oligonucleótidos antisentido. En un estudio más pequeño pero muy relevante que involucró tres pacientes con quilomicronemia familiar comprobada, volanesorsen indujo una profunda supresión de la producción de apoCIII, acompañada de reducciones en los TG plasmáticos de entre 56 y 86% 52. Por último, el estudio APPROACH fue un ensayo clínico aleatorizado, doble ciego, controlado con placebo, de 52 semanas duración y multicéntrico en 66 pacientes con quilomicronemia familiar, que evaluó la administración de volanesorsen 285 mg mediante inyección subcutánea (33 pacientes tratados con volanesorsen y 33 con placebo). Volanesorsen indujo una disminución de 84% en los niveles circulantes de apoC-III, lo que se tradujo en una reducción de los TG plasmáticos del 77% 53.
Con base en los resultados del estudio APPROACH, la Agencia Europea de Medicamentos (EMA), le otorgó en mayo de 2019 la aprobación a volanesorsen como terapia específica para la quilomicronemia familiar 50.
Evinacumab: anticuerpo monoclonal contra ANGPTL3
Como se mencionó en el segmento sobre el funcionamiento normal de la LPL-1, las ANGPTL son inhibidores endógenos de esta enzima, siendo ANGPTL3 la inhibidora más eficaz. Por esta razón, se han desarrollado anticuerpos monoclonales dirigidos contra ANGPTL3 como agentes reductores de triglicéridos. Evinacumab, actualmente en desarrollo clínico por Regeneron, es un anticuerpo completamente humano del isotipo IgG4, dirigido contra ANGPTL3. En evinacumab, la región bisagra de la molécula de IgG contiene una mutación estabilizadora para evitar que el agente conforme hemi-anticuerpos 54. En un primer estudio de prueba de concepto en 83 pacientes con TG entre 150 y 450 mg/dl, la administración intravenosa de evinacumab 20 mg/kg indujo una reducción del 76% en los TG plasmáticos, además de una reducción de 23% en el colesterol LDL. Por su parte la administración subcutánea indujo una reducción de TG de 64% en solamente 4 días, persistiendo hasta 15 días después de una única aplicación 55. El efecto adverso más frecuente con evinacumab fue cefalea en cerca de 10% de los pacientes. Actualmente se adelanta un estudio clínico de evinacumab en pacientes con hipertrigliceridemia >1000 mg/dl en al menos una oportunidad y al menos un evento de pancreatitis (NCT03452228), los resultados se esperan en la primera mitad del año 2021.
IONIS-ANGPTL3-LRx: oligonucleótido antisentido dirigido contra ANGPTL3
IONIS-ANGPTL3-LRx es un oligonucleótido antisentido de segunda generación conjugado a N-acetilgalactosamina y dirigido contra ANGPTL3. La conjugación con esta aminoazúcar hace al agente afín por el receptor 1 de asialoglicoproteínas en hepatocitos, lo que incrementa su selectividad por el hígado 56. La inhibición hepática de ANGPTL3 tiene un impacto ostensible sobre los lípidos plasmáticos: un estudio de prueba de concepto que incluyó a 44 pacientes con un amplio rango de triglicéridos plasmáticos, encontró que la administración semanal durante 6 meses de IONIS-ANGPTL3-LRx a dosis de 10, 20, 40, o 60 mg ocasionó reducciones de los TG plasmáticos entre 33 y 63%, con reducciones simultáneas de los niveles plasmáticos de apoB y apoC-III, sin eventos adversos serios 57.
Finalmente, debe mencionarse que existe el uso empírico del inhibidor de la proteína de transferencia microsomal de triglicéridos (MTTP-1) denominado lomitapida, aunque no se encuentra aprobado ni ha sido investigado específicamente para la indicación de quilomicronemia familiar 58.
Conclusiones
La quilomicronemia familiar es una enfermedad infrecuente pero relevante, con un profundo impacto médico y social en quienes la padecen. La aparición de nuevas estrategias terapéuticas que permitan atacar los defectos centrales manifiestos en esta enfermedad debe constituir un estímulo a la detección y tratamiento oportunos de los pacientes con esta enfermedad. La quilomicronemia familiar debe sospecharse, detectarse y tratarse oportunamente.
Agradecimientos: Agradecemos a la Universidad de los Andes por el apoyo logístico prestado para el desarrollo de este trabajo. Este trabajo fue apoyado por PTC Therapeutics
Conflicto de intereses: Ninguno para declarar
Bibliografía
1. Davidson M, Stevenson M, Hsieh A, et al. The burden of familial chylomicronemia syndrome: Results from the global IN-FOCUS study. J Clin Lipidol 2018; 12: 898-907.
2. Franssen R, Visser ME, Kuivenhoven JA, et al. Role of lipoprotein lipase in triglyceride metabolsim: potential therapeutic target. Future Lipidol 2008; 3: 385-97.
3. Kobayashi J1, Miyashita K, Nakajima K, et al. Hepatic lipase: a comprehensive view of its role on plasma lipid and lipoprotein metabolism. J Atheroscler Thromb 2015; 22: 1001-11.
4. Schönfeld P, Wojtczak L. Short- and medium-chain fatty acids in energy metabolism: the cellular perspective. J Lipid Res 2016; 57: 943-54.
5. Xiao C, Stahel P, Lewis GF. Regulation of chylomicron secretion: focus on post-assembly mechanisms. Cell Mol Gastroenterol Hepatol 2019; 7: 487-501.
6. Malloy M, Kane JP. Disorders of lipoprotein metabolism. En: Gardner DG, Shoback DM, editores: Greenspan’s basic & clinical endocrinology. Ohio, United States: Mc-Graw-Hill Education, 2017.
7. Kersten S. Angiopoietin-like 3 in lipoprotein metabolism. Nat Rev Endocrinol 2017; 13: 731-9.
8. Mendivil CO, Zheng C, Furtado J, Lel J, Sacks FM. Metabolism of very-low-density lipoprotein and low-density lipoprotein containing apolipoprotein C-III and not other small apolipoproteins. Arterioscler Thromb Vasc Biol 2010; 30: 239-45.
9. Lookene A, Beckstead JA, Nilsson S, et al. Apolipoprotein A-V-heparin interactions: implications for plasma lipoprotein metabolism. J Biol Chem 2005; 280: 25383-7.
10. Arora R, Nimonkar AV, Baird D, et al. Structure of lipoprotein lipase in complex with GPIHBP1. Proc Natl Acad Sci USA 2019; 116: 10360-5.
11. Shan L, Yu XC, Liu Z, et al. The angiopoietin-like proteins ANGPTL3 and ANGPTL4 inhibit lipoprotein lipase activity through distinct mechanisms. J Biol Chem 2009; 284: 1419-24.
12. Mehta N, Qamar A, Qu L, et al. Differential association of plasma angiopoietin-like proteins 3 and 4 with lipid and metabolic traits. Arterioscler Thromb Vasc Biol 2014; 34: 1057-63.
13. Robciuc MR, Tahvanainen E, Jauhiainen M, et al. Quantitation of serum angiopoietin-like proteins 3 and 4 in a Finnish population sample. J Lipid Res 2010; 51:824-31.
14. Mendivil CO, Rimm EB, Furtado J, et al. Low-density lipoproteins containing apolipoprotein C-III and the risk of coronary heart disease. Circulation 2011; 124: 2065-72.
15. Shachter NS. Apolipoproteins C-I and C-III as important modulators of lipoprotein metabolism. Curr Opin Lipidol 2001; 12: 297-304.
16. Schaap FG, Rensen PC, Voshol PJ, et al. ApoAV reduces plasma triglycerides by inhibiting very low density lipoprotein-triglyceride (VLDL-TG) production and stimulating lipoprotein lipase-mediated VLDL-TG hydrolysis. J Biol Chem 2004, 2; 279: 27941-7.
17. Young SG, Davies BS, Fong LG, et al. GPIHBP1: an endothelial cell molecule important for the lipolytic processing of chylomicrons. Curr Opin Lipidol 2007; 18:389-96.
18. Brahm A, Hegele RA. Hypertriglyceridemia. Nutrients 2013; 5: 981-1001.
19. Brahm AJ, Hegele RA. Chylomicronaemia-current diagnosis and future therapies. Nat Rev Endocrinol 2015; 11: 352-62.
20. Moulin P, Dufour R, Averna M, et al. Identification and diagnosis of patients with familial chylomicronaemia syndrome (FCS): Expert panel recommendations and proposal of an “FCS score.” Atherosclerosis 2018; 275: 265-72.
21. Gotoda T, Shirai K, Ohta T, et al. Diagnosis and management of type I and type V hyperlipoproteinemia. J Atheroscler Thromb 2012; 19: 1-12.
22. Hegele RA, Ginsberg HN, Chapman MJ, et al. The polygenic nature of hypertriglyceridaemia: implications for definition, diagnosis, and management. Lancet Diabetes Endocrinol 2014; 2: 655-66.
23. Nordestgaard BG, Freiberg JF. Clinical Relevance of Non-Fasting and Postprandial Hypertriglyceridemia and Remnant Cholesterol. Curr Vasc Pharmacol 2011; 9: 281-6.
24. Johansen CT, Hegele RA. Allelic and phenotypic spectrum of plasma triglycerides. Biochim Biophys Acta – Mol Cell Biol Lipids 2012; 1821: 833-42.
25. Dron JS, Wang J, Cao H, et al. Severe hypertriglyceridemia is primarily polygenic. J Clin Lipidol 2019; 13:80–8.
26. Chokshi N, Blumenschein SD, Ahmad Z, et al. Genotypephenotype relationships in patients with type I hyperlipoproteinemia. J Clin Lipidol 2014; 8: 287-95.
27. Patni N, Li X, Adams-Huet B, et al. The prevalence and etiology of extreme hypertriglyceridemia in children: Data from a tertiary children’s hospital. J Clin Lipidol 2018; 12: 305-10.
28. Paquette M, Bernard S, Hegele RA, et al. Chylomicronemia: Differences between familial chylomicronemia syndrome and multifactorial chylomicronemia. Atherosclerosis 2019; 283: 137-42.
29. Teramoto R, Tada H, Kawashiri M, et al. Molecular and functional characterization of familial chylomicronemia syndrome. Atherosclerosis 2018; 269: 272-8.
30. Wulff AB, Nordestgaard BG, Tybjærg-Hansen A. APOC3 Loss-of-Function Mutations, Remnant Cholesterol, Low-Density Lipoprotein Cholesterol, and Cardiovascular Risk: Mediation- and Meta-Analyses of 137 895 Individuals. Arterioscler Thromb Vasc Biol 2018; 38: 660-8.
31. Davidson M, Stevenson M, Hsieh A, et al. The burden of familial chylomicronemia syndrome: interim results from the IN-FOCUS study. Expert Rev Cardiovasc Ther 2017; 15: 415-23.
32. Blom D, O’Dea L, Digenio A, et al. Characterizing familial chylomicronemia syndrome: Baseline data of the APPROACH study. J Clin Lipidol 2018; 12: 1234-43.e5.
33. Pouwels E, Blom D, Firth J et al. Severe hypertriglyceridaemia as a result of familial chylomicronaemia: the Cape Town experience. S Afr Med J 2008; 98: 105-8.
34. Stroes E, Moulin P, Parhofer KG, et al. Diagnostic algorithm for familial chylomicronemia syndrome. Atheroscler Suppl 2017; 23: 1-7.
35. Hegele RA, Berberich AJ, Ban MR, et al. Clinical and biochemical features of different molecular etiologies of familial chylomicronemia. J Clin Lipidol 2018; 12: 920-7.
36. Otokozawa S, Ai M, Diffenderfer M, et al. Fasting and postprandial apolipoprotein B-48 levels in healthy, obese, and hyperlipidemic subjects. Metabolism 2009; 58: 1536-42.
37. Hegele R, Ban M, Cao H, et al Targeted next-generation sequencing in monogenic dyslipidemias. Curr Opin Lipidol 2015; 26: 103-13.
38. Johansen CT, Dubé JB, Loyzer MN, et al. LipidSeq: a nextgeneration clinical resequencing panel for monogenic dyslipidemias. J Lipid Res 2014; 55: 765-72.
39. Raffan E, Semple RK. Next generation sequencing–implications for clinical practice. Br Med Bull 2011; 99:53–71.
40. Fu J, Kwok S, Sinai L, Abdel-Razek O, et al. Western Database of Lipid Variants (WDLV): A Catalogue of Genetic Variants in Monogenic Dyslipidemias. Can J Cardiol 2013; 29: 934-9.
41. World Health Organization. Global recommendations on physical activity for health. Ginebra, Suiza, 2010. En: https://www.who.int/dietphysicalactivity/publications/9789241599979/en/; consultado diciembre 2019.
42. Connor WE, DeFrancesco CA, Connor SL. N-3 fatty acids from fish oil. Effects on plasma lipoproteins and hypertriglyceridemic patients. Ann N Y Acad Sci 1993; 14: 16-34.
43. Williams L, Rhodes KS, Karmally W, et al. Familial chylomicronemia syndrome: Bringing to life dietary recommendations throughout the life span. J Clin Lipidol 2018; 2: 908-19.
44. Ahmad Z, Wilson DP. Familial chylomicronemia syndrome and response to medium-chain triglyceride therapy in an infant with novel mutations in GPIHBP1. J Clin Lipidol 2014; 8: 635-9.
45. Elmadfa I, Kornsteiner M. Fats and fatty acid requirements for adults. Ann Nutr Metab 2009; 55: 56-75.
46. Helk O, Schreiber R, Widhalm K. Effects of two therapeutic dietary regimens on primary chylomicronemia in paediatric age: a retrospective data analysis. Eur J Clin Nutr 2016; 70: 1127-31.
47. Institute of Medicine. Panel on Dietary Reference Intakes for Electrolytes and Water. Dietary Reference Intakes: Water, Potassium, Sodium, Chloride, and Sulfate. Washington, D.C. United States: National Academies Press, 2004.
48. Toth P. Antisense therapy and emerging applications for the management of dyslipidemia. J Clin Lipidol 2011; 5: 441-9.
49. Crooke S. Molecular mechanisms of antisense oligonucleotides. Nucleic Acid Ther 2017; 27: 70-7.
50. European Medicines Agency. Waylivra (volanesorsen). En: www.ema.europa.eu/en/medicines/human/EPAR/waylivra; consultado julio 2019.
51. Gaudet D, Alexander VJ, Baker BF et al. Antisense Inhibition of apolipoprotein C-III in patients with hypertriglyceridemia. N Engl J Med 2015; 373: 438-47.
52. Gaudet D, Brisson D, Tremblay K, et al. Targeting APOC3 in the familial chylomicronemia syndrome. N Engl J Med 2014; 371: 2200-6.
53. Witztum JL, Gaudet D, Freedman SD, et al. Volanesorsen and triglyceride levels in familial chylomicronemia syndrome. N Engl J Med 2019; 381: 531-42.
54. Gusarova V, Alexa CA, Wang Y, et al. ANGPTL3 blockade with a human monoclonal antibody reduces plasma lipids in dyslipidemic mice and monkeys. J Lipid Res 2015; 56: 1308-17.
55. Dewey FE, Gusarova V, Dunbar RL, et al. Genetic and pharmacologic inactivation of ANGPTL3 and cardiovascular disease. N Engl J Med 2017; 377: 211-21.
56. Valencia-Enciso N, Mendivil CO. New biotechnological treatments for lipid disorders. Rev Invest Clin 2018; 70: 244-54.
57. Graham MJ, Lee RG, Brandt TA, et al. Cardiovascular and Metabolic Effects of ANGPTL3 Antisense Oligonucleotides. N Engl J Med 2017; 377: 222-32.
58. Brahm A, Hegele R. Lomitapide for the treatment of hypertriglyceridemia. Expert Opin Investig Drugs 2016; 25:1457-63.