
MARTA MARTINEZ-MORGA 1, CRISTINA MEDINA-CORVALAN 2, CLAUDIA PÉREZ-GARCÍA 2, CARLOS BUENO 1, SALVADOR MARTINEZ 2, 3
1 Departamento de Anatomía. Facultad de Medicina. Universidad de Murcia. IMIB-Arrixaca, 2 Cátedra de Neurociencia Aplicada. Universidad Católica de Murcia (UCAM). Murcia. 3 Instituto de Neurociencias UMH-CSIC. Alicante, España
Resumen Los trastornos heredados del metabolismo son enfermedades graves de la infancia que cursan con un gran deterioro cognitivo y del desarrollo psicomotor. La fisiopatología del progresivo deterioro del sistema nervioso suele estar asociada a una severa neuroinflamación y desmielinización, y como consecuencia, neurodegeneración. Por el momento no tienen cura y precisan de actitudes terapéuticas precoces y agresivas, que conllevan altas tasas de mortalidad y, muy frecuentemente, escasos grados de mejoría funcional y supervivencia. El trasplante de médula ósea y de células mesenquimales de médula ósea son terapias de elección y experimentales que consiguen mejorar el curso de estas enfermedades mediante diferentes mecanismos de acción: remplazo de enzima deficiente, intercambio de membranas y regulación del proceso inflamatorio..
Palabras clave: enfermedades lisosomiales de depósito, terapia celular, células mesenquimales, terapia de remplazo, corrección cruzada
Abstract Inherited metabolism disorders are serious childhood diseases that lead to significant cognitive impairment and regression of psychomotor development. The pathophysiology of the neural progressive deterioration is usually associated with severe neuroinflammation and demyelination, and as a consequence, neurodegeneration. At the moment they have no adequate treatment and require early and aggressive therapeutic approaches, which entail high mortality rates and, very frequently, low degrees of functional improvement and survival. Bone marrow transplantation and bone marrow mesenchymal cells grafts are therapeutic and experimental therapies that improve the course of these diseases through different mechanisms of action: enzyme replacement, membrane exchange and regulation of the inflammatory process.
Key words: deposit lysosomal diseases, cell therapy, mesenchymal cells, replacement therapy, cross-correction
e-mail: smartinez@umh.es
Las terapias avanzadas son nuevos desarrollos médicos que usan terapia génica, terapia celular e ingeniería de tejidos. Se pueden usar para tratar enfermedades o lesiones, como la piel en víctimas de quemaduras, enfermedades degenerativas, enfermedades congénitas por mutaciones genéticas y cáncer, y tienen un enorme potencial para el futuro de la medicina.
Las enfermedades hereditarias asociadas a mutaciones de genes conocidos están siendo tratadas con estrategias de remplazo (ER) del producto codificado por el gen alterado. En aquellas enfermedades en las que el producto del gen puede ser producido por células de la médula ósea (MO) y transferido a otras células, el trasplante alogénico de médula ósea (TMO) es muchas veces una solución terapéutica 1. Cuando no se encuentra el donante, se puede acudir a terapia génica (TG) y modificar genéticamente células del propio paciente: trasplante autólogo con TG 2.
Los trastornos hereditarios del metabolismo representan un grupo diverso y complejo de enfermedades causadas por defectos en los genes que codifican proteínas funcionales de las vías metabólicas. El TMO es una opción terapéutica, en ocasiones como primera línea de elección, para estas enfermedades, donde otras terapias disponibles son menos efectivas, y donde el beneficio del TMO supera el riesgo del trasplante 1.
Este artículo revisa y discute los mecanismos de la terapia celular en ciertas enfermedades lisosomiales y peroxisomiales de depósito.
Enfermedades lisosomiales de depósito
Fisiopatología
Las enfermedades lisosomiales de depósito (ELD) son un grupo heterogéneo de patologías producidas por mutaciones en genes que codifican proteínas específicas de los lisosomas. La función de los lisosomas se ha estudiado mucho estos últimos años y se ha visto su implicación en otros procesos más allá de su papel en la degradación y el reciclaje de productos celulares.
Así, sabemos que desempeñan un papel crucial en la reparación de la membrana plasmática, el intercambio de lípidos y metabolitos entre los orgánulos celulares.
También que regulan el metabolismo energético a través de la señalización de calcio 3. La patogénesis de la lesión celular en las ELD no es bien conocida, pero se debe en parte a la acumulación primaria de sustratos no digeridos y mal procesados dentro de los lisosomas y a la posterior patología derivada de estos depósitos. Esta disfunción en el manejo de sustratos parece debida fundamentalmente a la alteración de los procesos regulados por proteínas de la pared de los lisosomas; proteínas que son necesarias para la maduración funcional de los procesos de digestión y reciclaje de residuos celulares.
Indicación para TMO en ELD
Las ELD requieren intervención precoz, para evitar el daño irreversible de los tejidos mas afectados; por lo tanto, deben tratarse en fases asintomáticas o al inicio de los síntomas y abordarse desde un manejo multidisciplinar, para optimizar la respuesta al tratamiento, la calidad de vida después de este y prevenir la mortalidad prematura.
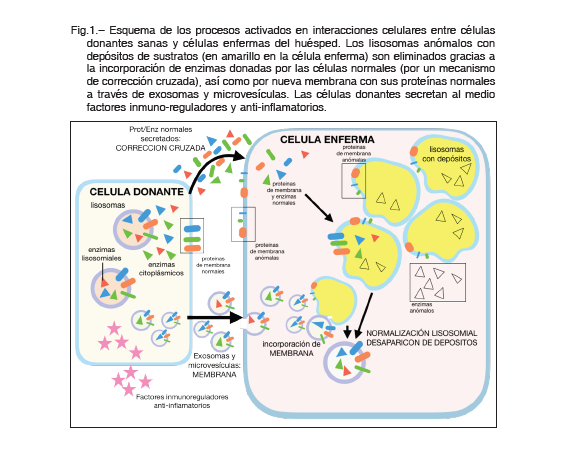
El mecanismo de acción del TMO en las ELD se conoce como corrección cruzada (Figura 1). Es decir, las células trasplantadas proporcionan a las células del receptor una fuente continua de enzimas. Estas enzimas son producidas por células mieloides derivadas de la MO de donantes sanos, que luego son absorbidas por las células el huésped que son deficientes en ellas 4. Además, la superioridad del TMO con respecto a la terapia de reemplazo mediante administración farmacológica de las enzimas, en relación a la afectación del neural, radica en la capacidad de las células derivadas de la MO donante para migrar a través de la barrera hematoencefálica y diferenciarse en microglía. Esta microglía donante secreta la enzima deficiente al espacio extracelular del parénquima del sistema nervioso central (SNC) y mejora la afectación neural, y con ello los resultados neurocognitivos. Las mucopolisacaridosis (MPS) son el paradigma del éxito del TMO en la enfermedad metabólica. El TMO es de elección para el tratamiento en pacientes con MPS II (síndrome de Hurler) que son menores de 2 años, que no tienen o tienen un deterioro cognitivo mínimo 5.
La mucopolisacaridosis tipo 1 (MPS I) es una ELD recesiva caracterizada por el depósito de mucopolisacáridos o glucosaminoglucanos (GAG) en los lisosomas. Los pacientes tienen una mutación defectuosa en el gen IDUA que codifica la alfa-L-iduronidasa, produciendo un catabolismo ineficaz de heparán y dermatán sulfatos. Estos depósitos de GAG en los órganos provocan una disfunción multiorgánica, que aparece como un espectro clínico de gravedad variable, con retraso mental progresivo, deformidades esqueléticas, enfermedad gastrointestinal y discapacidad visual y auditiva. El síndrome de Hurler, es el fenotipo más grave de MPS con una enfermedad de inicio temprano y de progresión rápida con afectación neurológica.
Los tratamientos farmacológicos de remplazo enzimático disponibles actualmente no previenen el deterioro cognitivo, ya que no cruzan la barrera hematoencefálica en dosis suficientes. Además, la terapia farmacológica a largo plazo está limitada por la inducción de anticuerpos antienzimáticos, lo que disminuye la reducción del sustrato 6.
Esfingolipidosis
Fisiopatología
La Enfermedad de Niemann-Pick (ENP) es una enfermedad hereditaria autosómica recesiva que consiste en una anormalidad en el metabolismo de los lípidos, proceso que se da en el interior de los lisosomas y que conlleva a una acumulación de sustratos lipídicos y colesterol en el interior de los mismos. Clínicamente se caracteriza por aparición de síntomas neurológicos progresivos, como consecuencia del daño cerebral. Existen dos formas de presentación, la infantil o precoz donde los síntomas aparecen relativamente pronto, antes de los dos años y con un mal pronóstico; y la forma juvenil, en la que los síntomas aparecen después de los 10 años y con mejor pronóstico (siendo en ambos casos progresiva e irrecuperable). Se conocen tres tipos de ENP: A, B y C. La ENP tipo C (ENPC) se manifiesta con un marcado deterioro neurológico y en menor medida con una afectación hepato-esplénica. Pertenece a un grupo muy heterogéneo de lipidosis en las que, aunque la presentación y diagnóstico suele producirse en la infancia, existe un amplio espectro de formas de presentación en edades posteriores. Por ello, clínicamente el rango va desde un rápido y fatal desarrollo en neonatos a una neurodegeneración crónica en adultos, con ataxia cerebelosa, disfagia, demencia y muerte. Este tipo de ENP es causado por mutaciones tanto en el gen NPC1 como en el NPC2, que codifican proteínas de la membrana de los lisosomas. Por lo tanto, estas mutaciones conducen a defectos en el transporte del colesterol en el lisosoma y su acumulación en las células del SNC y periférico (tanto en neuronas como glía). El diagnóstico se establece mediante la detección de estas anomalías en cultivos de fibroblastos. El diagnóstico prenatal se obtiene de manera más fácil mediante técnicas de biología molecular, pero también se puede llevar a cabo mediante técnicas de biología celular.
Hasta la fecha, no hay un tratamiento específico disponible. Se han obtenido resultados prometedores en modelos animales (gato y ratón) con un inhibidor de la síntesis de glucolípidos, lo que ha dado lugar al inicio de un ensayo clínico. El pronóstico depende de la edad de inicio de las manifestaciones neurológicas, más grave en los casos en los que la afectación neurológica es de aparición temprana (información: https://www.orpha.net).
Enfermedades peroxisomiales de depósito
Fisiopatología
La adrenoleucodistrofia ligada al X (X-ALD) con localización cerebral (X-CALD), es una enfermedad peroxisomal caracterizada por una desmielinización inflamatoria grave en el cerebro y a menudo asociada a insuficiencia suprarrenal. La X-CALD puede producirse en niños varones sanos (2.5-10 años de edad, ~50% de casos), en casos de adreno-mieloneuropatía (AMN) en varones sintomáticos (35%), en adultos varones como la manifestación inicial del X-ALD (~12%) y con mucha menor frecuencia en mujeres adultas (~2%). La insuficiencia suprarrenal (IS, ~65% de casos) es a menudo latente, con ausencia de melanodermia, presentándose como fatiga, náusea o incluso insuficiencia suprarrenal aguda primaria. Le sigue una fase activa con: labilidad emocional, declive cognitivo y alteraciones visuo-espaciales; así como síndromes frontales a los que se les unen rápidamente déficits motores como hemiplejía o tetraparesia, ataxia cerebelosa, deterioro de la discriminación auditiva central, defectos del campo visual, ceguera cortical y convulsiones. La X-ALD se debe a mutaciones en el gen ABCD1 (Xq28) que codifica para la ALDP, una proteína transmembrana peroxisomal implicada en el transporte del citosol al peroxisoma de ésteres-CoA de ácidos grasos de cadena muy larga (VLCFA). La resonancia magnética cerebral (RM) es el único medio para detectar la desmielinización cerebral antes de la aparición de los síntomas, o incluso con déficits cognitivos leves. En varones con X-ALD con una RM cerebral normal, la RM debe repetirse cada 6 meses entre los 3-12 años, y a partir de entonces anualmente hasta los 50 años.
El curso clínico de la enfermedad y la falta de signos de remielinización en estudios neuropatológicos y de resonancia magnética, indican que las capacidades de regeneración de la mielina están obstaculizadas, debido a razones que actualmente no se conocen. Curiosamente, todos los fenotipos clínicos pueden ocurrir dentro de la misma familia e incluso ser discordantes en gemelos monocigóticos; es decir, no existe una correlación fenotipo-genotipo. Por lo tanto, se supone que existe la participación de factores epigenéticos en la expresividad variable de la enfermedad 7.
El TMO solo está indicado en la forma de afectación cerebral (X-CALD) y es el único tratamiento disponible para mejorar el curso de la enfermedad. Es poco probable que la eficacia del TMO en X-ALD esté solo en la corrección cruzada, por lo que se ha pensado que otros factores de la enfermedad, como el neurotrofismo y un efecto antinflamatorio, están siendo la diana de este tratamiento.
El TMO puede detener el proceso de desmielinización al disminuir el proceso inflamatorio, regular el sistema inmune y reemplazar la microglía disfuncional con macrófagos derivados de la médula ósea. Esta multiplicidad de mecanismos de acción puede explicar de forma individual y conjunta la actividad terapéutica de la terapia celular.
Indicación actual de terapia celular en trastornos heredados del metabolismo
La investigación sobre enfermedades neurodegenerativas ha puesto en evidencia dos mecanismos que están especialmente presentes en los trastornos hereditarios del metabolismo asociados a la disfunción de los lisosomas. Estos mecanismos tienen que ser adecuadamente conocidos a la hora de buscar nuevos tratamientos de terapias avanzadas:
1) Terapia celular usando las células como biofármacos: durante los últimos 30 años, y especialmente en la última década, se ha postulado la posibilidad de utilizar células mesenquimales de MO o de cordón umbilical (CMMO) como una alternativa terapéutica al TMO. Experimentos en modelos animales de enfermedad han emostrado que las CMMO son capaces de ralentizar, detener o incluso regenerar daños en los tejidos. Esto es debido a la vasta variedad de mecanismos de señalización celular que estas células pueden llevar a cabo, tales como la capacidad de fusión celular, liberación continuada de factores moleculares funcionalmente activos (incluida la corrección cruzada), inmunomodulación y la activación de las células progenitoras de los órganos donde se implantan 8. Paralelamente, experimentos de co-cultivo de células mesenquimales, de diferentes orígenes, con células neurales han descubierto la capacidad de las células mesenquimales para producir vesículas extracelulares (exosomas y microvesículas) que pueden transportar elementos tróficos y beneficiosos a las células con las que se fusionan, de acuerdo con las conclusiones del reciente documento de revisión publicado por Phinney y Pittenger 9. Pero, lo que parece más importante para ayudar a las células a eliminar depósitos de sustancias tóxicas es proporcionarles una membrana celular abundante y normal (portadora de proteínas deficientes de membrana características de estas enfermedades), con la posibilidad de introducir receptores y activar señales lisosomiales 10,11 (Figura 1). En nuestro laboratorio hemos aislado células mesenquimales de la pulpa dental, mostrando características de las células madre de la cresta neural y, por lo tanto, pueden generar neuronas en cultivos neurogénicos. Con cultivos adecuados, estas células de pulpa dental permiten el desarrollo de modelos de enfermedades genéticas humanas para estudios in vitro, en nuestro caso la enfermedad estudiada fue la ataxia de Friedreich, demostrando el benéfico terapéutico de las células mesenquimales (en co-cultivos celulares) y sus sobrenadantes (en cultivos aislados) para la expresión del gen de la frataxina 12,13.
2) Además de depósito en los lisosomas de sustratos mal procesados, las ELD se acompañan de un proceso de neuroinflamación muy importante, con disfunción del sistema inmune. Cada día está más claro la implicación de un proceso subyacente de neuroinflamación para entender la patogenia de estas enfermedades. En este sentido la pérdida de integridad de la barrera hematoencefálica y la activación de la inmunidad innata nos aparecen como elementos patogénicos importantes para entender el proceso de inflamación crónica y para ser estudiado como posible diana terapéutica. Es sorprendente admitir los pocos estudios sobre el papel de las células de la unidad neuro-vascular en estas enfermedades y su potencialidad como diana terapéutica 14, 15.
La heterogeneidad clínica de las ELD, y en general los trastornos heredados del metabolismo, indica que además de factores genéticos, que son los desencadenantes del trastorno funcional, existen otros procesos derivados de mecanismos reactivos regulados por factores externos e internos que mantienen la progresión y el agravamiento de la enfermedad. La aproximación terapéutica ha de intentar incidir sobre el mayor número de factores alterados; por ejemplo, en el caso de la X-CALD, ha de actuarse sobre la inflamación, desmielinización y activación de progenitores oligodendrogliales para reactivar la remielinización. Las células, actuando como bio-fármacos, tiene la capacidad de actuar sobre estos procesos de forma eficiente. Tienen propiedades neurotróficas e inmunoreguladoras, además de activar localmente los progenitores indiferenciados. Creemos que la terapia celular puede representar en un futuro próximo una vía terapéutica por estas enfermedades, que por el momento carecen de ella.
Conflicto de intereses: Ninguno para declarar
Bibliografía
1. Tan EY, Boelens JJ, Jones SA, Wynn RF. Hematopoietic stem cell transplantation in inborn errors of metabolism. Front Pediatr 2019; 7: 433, eCollection 2019.
2. Staal FJT, Aiuti A, Cavazzana M. Autologous stem-cellbased gene therapy for inherited disorders: State of the art and perspectives. Front Pediatr 2019; 7: 443.
3. Medina DL, Ballabio A. Lysosomal calcium regulates autophagy. Autophagy 2015; 11: 970-1.
4. Bonam SR, Wang F, Muller S. Lysosomes as a therapeutic target. Nat Rev Drug Discov 2019; 18: 923-48.
5. Muenzer J, Jones SA, Tylki-Szymańska A, et al. Ten years of the Hunter Outcome Survey (HOS): insights, achievements, and lessons learned from a global patient registry. Orphanet J Rare Dis 2017; 12: 82.
6. Dickson P, Peinovich M, McEntee M, et al. Immune tolerance improves the efficacy of enzyme replacement therapy in canine mucopolysaccharidosis I. J Clin Invest 2008; 18: 2868-76.
7. Schlüter A, Sandoval J, Fourcade S, et al. Epigenomic signature of adrenoleukodystrophy predicts compromised oligodendrocyte differentiation. Brain Pathol 2018; 28: 902-19.
8. Watanebe, TK. A Review of Stem Cell Therapy for Acquired Brain Injuries and Neurodegenerative Central Nervous System Diseases. PM-R 2018; 10 (Suppl 2): S151-6.
9. Phinney DG, Pittenger MF. Concise review: MSC-derived exosomes for cell-free theraphy. Stem Cells 2017; 35: 851-8.
10. Schneider JL, Cuervo AM. Autophagy and human disease: merging themes. Curr Opinion Genet Dev 2014; 26: 16-23.
11. Koniusz S, Andrzejewska A, Muraca M, Srivastava AK, Janowski M, Lukomska B. Extracellular vesicles in physiology, pathology, and therapy of the immune and central nervous system, with focus on extracellular vesicles derived from mesenchymal stem cells as therapeutic tools. Front Cell Neurosci 2016; 10: 109.
12. Jones J, Estirado E, Redondo C, Martinez S. Human adipose stem cells rescue Friedreich’s ataxia cells from oxidative stress and increase frataxin expression. Stem Cells Dev 2012; 21: 2817-26.
13. Jones J, Estirado E, Redondo C, Martinez S. Stem cells from wildtype and Friedreich’s ataxia mice present similar neuroprotective properties in dorsal root ganglia cells. PLoS One 2013; 8(5): e62807.
14. Pombero A, Garcia-Lopez R, Martinez S. Brain mesenchymal stem cells: physiology and pathological implications. Dev Growth Differ 2016; 58: 469-80.
15. Wilson JJ, Foyle KL, Foeng J, et al. Redirecting adult mesenchymal stromal cells to the brain: a new approach for treating CNS autoimmunity and neuroinflammation? Immunol Cell Biol. 2018; 96: 347-57.