*en inglés
DIEGO RIPEAU ¹, HERNÁN AMARTINO ², MARTÍN CEDROLLA ³, LUIS URTIAGA ³, BELLA URDANETA 4, MARILIS CANO 5, RITA VALDEZ 6, NORBERTO ANTONGIOVANNI 7, FRANCISCA MASLLORENS ¹
¹ Centro de Estudio de Enfermedades Lisosomales, Hospital Nacional Prof. Dr. Alejandro Posadas, Haedo, Argentina, ² Fundación Investigar, Buenos Aires, Argentina, ³ Hospital Regional Ángela Llano, Corrientes, Argentina, 4 Hospital de San José de Buga, Guadalajara, Colombia, 5 Nefrología, Hospital General del Sur Dr. Pedro Iturbe, Maracaibo, Venezuela, 6 Hospital Militar Central, Servicio de Genética, Buenos Aires, Argentina, 7 Instituto de Nefrología Pergamino, Pergamino, Argentina
Resumen Cambio de agalsidasa beta por agalsidasa alfa en la terapia de reemplazo enzimática de pacientes con enfermedad de Fabry en Latinoamérica. Actualmente hay disponibles dos terapias de reemplazo enzimático en enfermedad de Fabry y existe poca información sobre la eficacia y seguridad del cambio de una a la otra. Entre 2009 y 2012 hubo falta a nivel mundial de agalsidasa beta y los pacientes tratados hasta entonces con esa enzima iniciaron tratamiento con agalsidasa alfa. El presente estudio retrospectivo, observacional evaluó la eficacia y seguridad a 2 años en pacientes con enfermedad de Fabry en Argentina (30 pacientes) y Venezuela (3 pacientes), que cambiaron su tratamiento de agalsidasa beta a agalsidasa alfa. Treinta y tres pacientes completaron 24 meses de seguimiento post-cambio (edad 32.4 ± 2.0; rango 10.0-55.9; hombre: mujer 23:10). La función renal, medida con la tasa de filtrado glomerular, se mantuvo sin cambios en 31 pacientes sin enfermedad renal terminal durante 2 años post-cambio. La secreción de proteína en orina continuó estable. Los parámetros de función cardíaca –índice de masa ventricular izquierda, septum interventricular, espesor de la pared posterior ventricular– no mostraron cambios significativos post-cambio de terapia en los 33 pacientes. La calidad de vida, el dolor y la gravedad de la enfermedad se mantuvieron mayormente estables luego de 24 meses, y la agalsidasa alfa fue generalmente bien tolerada. Nuestros resultados muestran que no hay cambios significativos en la eficacia medida por la función renal y cardíaca, en la seguridad y en los valores de la calidad de vida, el dolor o la gravedad de la enfermedad durante al menos 2 años luego del cambio de agalsidasa beta a agalsidasa alfa.
Palabras clave: enfermedad de Fabry, agalsidasa alfa, agalsidasa beta, cambio
Abstract There are currently two available enzyme replacement therapies for Fabry disease and little information regarding efficacy and safety of switching therapies. Between 2009 and 2012 there was a worldwide shortage of agalsidase beta and patients on that enzyme were switched to agalsidase alfa. This retrospective observational study assessed a 2-year period of efficacy and safety in a population of Fabry patients, in Argentina (30 patients) and Venezuela (3 patients), who switched therapies from algasidase beta to agalsidase alfa. Thirty-three patients completed 24-months follow-up after the switch (age 32.4 ± 2.0, range 10.0-55.9 years; male: female 23:10). Measures of renal function such as estimated glomerular filtration rate remained almost unchanged in 31 patients without end stage renal disease over the 2 years after switching and urine protein excretion continued stable. Cardiac functional parameters: left ventricular mass index, interventricular septum, left ventricular posterior wall showed no significant change from baseline in the 33 patients. Quality of life, pain and disease severity scores were mostly unchanged after 24-months and agalsidase alfa was generally well tolerated. Our findings showed there is no significant change in the efficacy measured through the renal or cardiac function, quality of life, pain, disease severity scoring and safety for at least 2 years after switching from agalsidase beta to agalsidase alfa.
Key words: Fabry disease, agalsidase alfa, agalsidase beta, switch
Received: 22-VIII-2016 Accepted: 13-III-2017
Postal address: Francisca Masllorens, Centro de Estudio de Enfermedades Lisosomales, Hospital Nacional Prof. Dr. Alejandro Posadas, Av. Marconi y Pte. Illia, 1706 Haedo, Buenos Aires, Argentina.
e-mail: franmasllorens@yahoo.com.ar
Fabry disease (FD) is a progressive, multisystemic and devastating X-linked lysosomal storage disorder caused by reduced or absent activity of the hydrolytic enzyme α-galactosidase A (α-Gal A)1. The deficiency of this enzyme results in the accumulation of several neutral glycolipids mainly globotriaosylceramide (GL-3) in lysosomes of fibroblasts, endothelial cells, pericytes from dermal tissue, heart, kidney, and autonomic nervous system, triggering inflammation and fibrosis and progressive vital organ dysfunctions2-4. FD affects the heart, kidneys, and cerebrovascular system, which contribute to a reduced quality of life and life expectancy5.
The current treatment for FD is intermittent intravenous administration of the deficient lysosomal enzyme6-10.
Two different α-galactosidase A treatments have been approved for the treatment of FD: agalsidase alfa (Replagal®; Shire Human Genetic Therapies, Lexington, MA) and agalsidase beta (Fabrazyme®; Genzyme, Cambridge, MA). As of the writing of this manuscript, thousands of patients have been treated with these two enzyme replacement therapies (ERTs) worldwide11. There is only anecdotal data regarding the comparative efficacy and safety between these two agents12, 13.
As from mid-2009 a viral contamination in the agalsidase beta production resulted in a worldwide scarcity of agalsidase beta. A consensus emerged that switching to agalsidase alfa at 0.2 mg/kg EOW was a plausible option14. This represents a unique circumstance in which a large number of patients switched treatments for reasons other than lack of efficacy.
In this paper we report the main clinical findings and the outcome of a cohort of 33 Latin American patients with FD who were receiving long-term agalsidase beta and had to be switched to agalsidase alfa due to manufacturer´s shortage occurred between 2009 and 2012 of the former.
The aim of this study was to evaluate changes in renal function and cardiac imaging up to 24 months’ post switch. Secondary objectives were to assess the safety and clinical changes when switching ERTs.
Materials and methods
This was a multicenter, retrospective, chart review, observational study of FD individuals who switched from treatment with agalsidase beta (1.0 mg/kg EOW) to agalsidase alfa (0.2 mg/kg EOW), conducted in two Latin American countries. A total of 33 patients were included: 30 patients from Argentina and 3 patients from Venezuela.
This trial was carried-out according to the Declaration of Helsinski and other local regulations and was approved by an Institutional Review Board and Ethic Committee. All the participants signed the appropriate written informed consent.
Individuals included in the analysis were hemizygous males or heterozygous females of any age with genetically confirmed diagnosis of FD, had been receiving agalsidase beta prior to starting treatment with agalsidase alfa in the recommended dose (i.e. 1.0 mg/kg ± 10% every other week [EOW]) or at least 12 months ± 2 and provided a written consent. Patients were excluded if there was concomitant use of agalsidase alfa, if the switch occurred before 2009 or if it was done for lack of efficacy reasons, or if agalsidase beta was administered at reduced dose for more than 6 months.
In addition to ERT patients received supportive medications like angiotensin-converting enzyme inhibitors (ACEIs), angiotensin receptor antagonists (ARAs), beta blockers or dual adrenergic blockers, vasodilators or any other blood pressure lowering agent as well as any other symptoms relieving drugs. In order to reduce infusion-associated reactions, patients receiving pretreatment with histamine receptor antagonists or non-steroidal anti-inflammatory drugs continued with the same regimen after swicht.
The following medical evaluations were recorded at baseline (ERT switching) and at 12-month intervals after switching from agalsidase beta to agalsidase alfa: clinical examination, complete blood count (CBC), blood chemistries, and urinalysis; serum creatinine and estimated glomerular filtration rate (e-GFR) calculated through the Chronic Kidney Disease Epidemiology Collaboration equation (CKD-EPI equation)15; urine protein excretion following 24 hour urine collection and the urine protein/creatinine ratio; a 12-lead ECG and bi-dimensional echocardiography (EKG) were used to estimate interventricular septum (IVS) left ventricular wall thickness, left ventricular mass (LVM) and left ventricular ejection fraction (LVEF); health status was evaluated with the Short Form-36 (SF-36); pain score, assessed with the Brief Pain Inventory (BPI) and the severity of Fabry disease signs and symptoms using the Mainz Severity Score Index (MSSI).
The primary endpoints were the absolute change in eGFR and the change in echocardiographic parameters (septum and posterior wall thickness, left ventricular (LV) size in diastole and systole, left atrial size, filling impairment, ejection fraction, valvular lesions) from the time of switch to 2 years post switch. The secondary endpoints were change in other quantifiable clinical and laboratory parameters from switch and up to 2 years post switch, including 24 hour urinary protein, protein/creatinine ratio, change in BPI (average and worst score), SF-36, MSSI from switch and 2 years post switch and occurrence of adverse events, change in vital signs and use of concomitant medications.
In order to assess the safety of agalsidase alfa all adverse events (AEs) were recorded; including their severity and relationship to the study drug.
All data are expressed as mean ± SEM, median or proportions unless otherwise indicated. Normal distribution of the study variables was assessed through the Shapiro-Wilks test. The Levine’s and the Mauchly’s tests were used to corroborate homogeneity of variances and sphericity when appropriated. To assure normal distribution some variables (eGFR and urine protein excretion or the protein/creatinine ratio) were log transformed. Repeated measures ANOVA were used to test the effect of treatment at any time point (switch, 12 and 24 months) and the non-parametric Wilcoxon Mann-Whitney test was used to evaluate changes in those variables with non-Gaussian distribution. All tests are two tailed and values of p < 0.05 were considered statistically significant.
Results
Thirty-three (n = 33) FD patients (23 males aged 29.4 ± 2.31 range 10.0-51.5 and 10 females aged 39.4 ± 3.01 range 25.1-55.9) years who met the inclusion criteria were included in the analysis. Four patients were less than eighteen years of age at the time of switch. Follow up data at 24 months was available for all patients.ENZYME REPLACEMENT SWITCH FROM AGALSIDASE BETA TO AGALSIDASE ALFA 175
Age at diagnosis of FD was 26.7 ± 2.1 years (range 4.5-53.1 years) and age at start of ERT with agalsidase beta was 28.7 ± 2.1 years (5.7-55.0 years). Average ERT treatment time before switching was 43.7 ± 5.4 months. Mean age at the time of switching was 32.4 ± 2.0 years.
Patients presented different mutations the most common (n = 14) being a mutation in the L415P encoding gene. Other previously described, as well as novel mutations were equally distributed within this cohort.
The main baseline (at switch) and follow-up characteristics of this cohort is shown in Table 1.
At the time of switching 22 patients had normal eGFR (> 90 mL/min/1.73 m2); 7 additional individuals had stage 2 CKD; two had stage 3 CKD and two had stage 5 CKD and were on renal replacement therapy.
The two stage 5 CKD patients requiring hemodialysis were 44 and 47 year-old males. They had no cause of CKD other than FD, but kidney biopsies were not performed. They were diagnosed with FD at age of 29 and 45 respectively and had been on ERT for 8 and 1 years before switching. They were normotensive and, at the time of switching, both displayed evidence of moderate-severe heart involvement. These patients were excluded from the eGFR endpoint analysis.
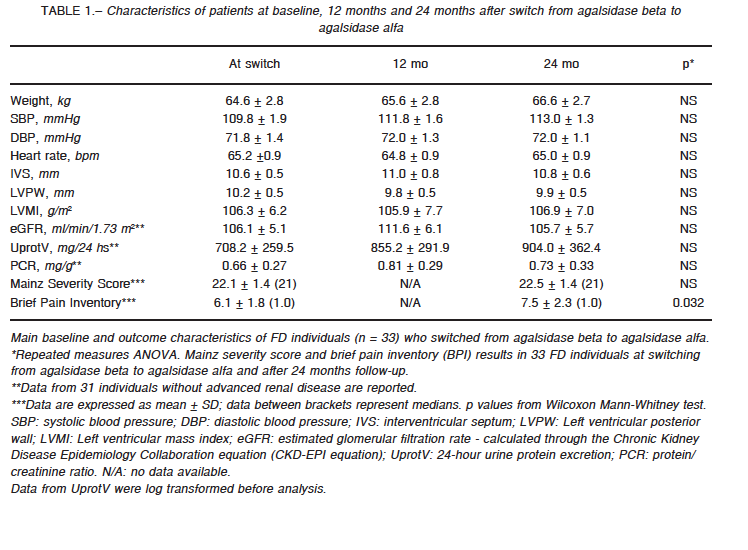
eGFR was assessed through the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equation; 24-hour urine protein excretion and protein-creatinine ratio was evaluated at baseline and yearly for 24 months in those 31 FD individuals without end stage renal failure receiving agalsidase alfa. eGFR (ml/min/1.73 m2) remained largely unchanged; from 106.1 ± 5.1 (baseline) to 111.6 ± 6.1 and 105.7 ± 5.7 at 12 and 24 months respectively (p: NS) (Fig. 1a). Similarly, and despite the statistically non-significant stepwise tendency to increase, urine protein excretion (mg/24h) remained stable (708.2 ± 259.5; 855.2 ± 291.9; and 904.0 ± 362.4 at baseline, and at 1 and 2 years respectively (p = NS) (Fig. 1b).
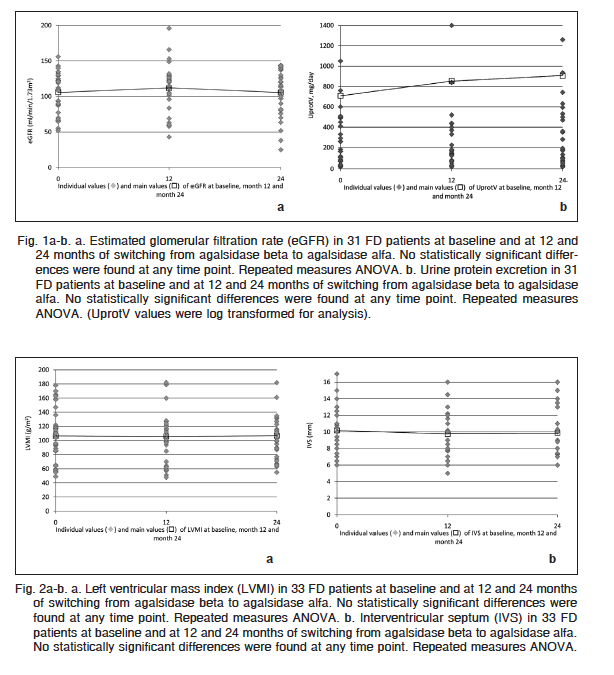
Parameters of cardiac structure and function remained stable after the treatment switch (Table 1). LVMI was similar at switch (106.3 ± 6.2 g/m2) and 12 months (105.9 ± 7.7 g/m2) and 24 months (106.9 ± 7.0 g/m2) after switching (p: NS) (Fig. 2a). Similarly, there was no significant change in IVS (mm) measured by cardiac ultrasound from baseline (10.6 ± 0.5) at 12 (11.0 ± 0.8) and 24 months of agalsidase alfa (10.8 ± 0.6) (Fig. 2b).
LVPW remained unchanged throughout the entire follow-up period (10.2 ± 0.5 mm at baseline) compared to 9.8 ± 0.5 and 9.9 ± 0.5 mm at 12 and 24 months after switch (p: NS). There were no statistically significant differences between baseline and 12 and 24 months SBP (109.8 ± 1.9; 111.8 ± 1.6 and 113.0 ± 1.3 mmHg respectively – p: NS) or DBP (71.8 ± 1.4; 72.0 ± 1.3 and 72.0 ± 1.1 mmHg respectively – p: NS).
Heart rate remained unchanged from 65.2 ± 0.9 bpm (at switch) to 64.8 ± 0.9 (12 months) and to 65.0 ± 0.9 (24 months) bpm after switching (p: NS). Similarly, no clinically significant EKG changes were observed during the 24 months follow-up after switching.
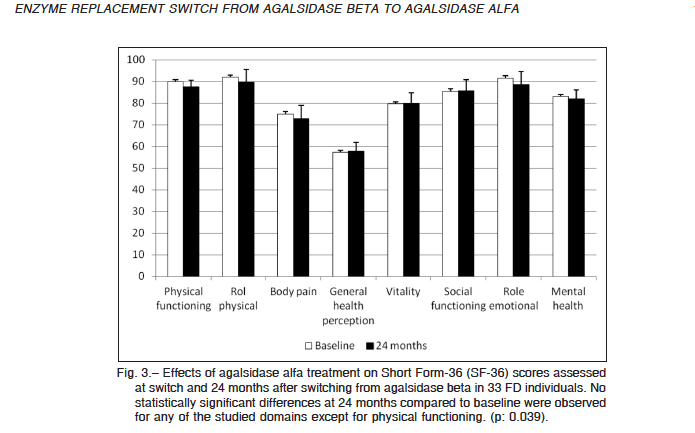
All the enrolled patients had the SF-36 self-assessment questionnaire completed at switch (baseline) and after 24 months of treatment with agalsidase alfa. The overall score remained unchanged, as well as the majority of the eight domains included in the score. Change was 7 points or less for all eight domains (Fig. 3).
The physical functioning component of the SF36 form was the single domain that experienced a statistical significant change in scoring, from 89.9 ± 2.3 (median 95.0) points at baseline to 87.7 ± 2.6 (median 95.0) after 24 months of agalsidase alfa (p = 0.039). A single 46-year-old male individual showed a significant worsening in all of the subscales of the SF36 questionnaire.
The overall physical health summary remained unchanged in 16 individuals, improved in 9 and worsened in 8 (p: NS); similarly, the mental health summary persisted invariable in 18, improved in 9 but deteriorated in 6 (p = NS).
Pain intensity was separately assessed through the Brief Pain Inventory (BPI) questionnaire. A statistically but not clinically significant increase of 22% in BPI (from baseline of 6.09 + 1.8 -median: 0 to 7.7 + 2.3 – median: 0 at 24 months post switch) was observed in this cohort of 33 individuals after 24 months of switching to agalsidase alfa (p: 0.032) (Table 1).
Pain intensity remained unchanged in 82% of the individuals (n = 27); improved in 2 and worsened in 4.
Two of the 4 patients who had worsening of pain score presented with carpal tunnel syndrome and the reported pain was specifically in hands. Carpal tunnel syndrome was confirmed by biopsy and the pain resolved completely after surgery.
The MSSI showed that the participants presented mild manifestations of FD at baseline and that the disease remained stable 24 months after switching to agalsidase alfa (Table 1).
Physical examination along with blood tests had been performed regularly throughout the entire period of observation. No treatment-related clinical or laboratory adverse reactions were noted. No treatment-associated arrhythmias or heart failure were reported within 24h of administration of ERT.
Discussion
The number of communications about the effect of switching from agalsidase beta to agalsidase alfa is increasing. However, there is limited amount of data available in the literature on Latin American patients16. To our knowledge, this is the first multicenter, binational study in a population of FD patients evaluating the effects of switching agalsidase beta to agalsidase alfa. Renal and cardiac function, quality of life, pain symptoms and disease severity were mostly unchanged in the 33 patients with FD switched from ERT after 24 months, suggesting patients maintained disease stability. The treatment switch was also generally well tolerated.
Our results largely correspond with those described by Japanese17, Taiwanese13 and Italian groups18. Tsuboi et al reported a 3-year follow-up of 11 Japanese FD patients switched from agalsidase beta to agalsidase alfa. Vital organ function (cardiac and renal) remained almost unaffected after switching17. Lin et al reported a 1-year post-switch follow up of 9 FD patients showing stability in renal function, cardiac parameters and MSSI scores. They also have investigated lysoGb3 plasma levels with no significant change13. Pisani et al in 10 FD patients switched over from agalsidase beta to algasidase alfa and followed during 20 months found no significant changes in renal function or cardiac functional parameters, pain symptoms or health status, and the switch was generally well tolerated. Patients were able to maintain a clinically stable disease state after the switch to long-term agalsidase alfa treatment18.
The results of the current study are also consistent with those of Smid et al. They retrospectively investigated the effects of the agalsidase beta shortage in 35 patients with FD disease in The Netherlands19. Twenty patients were switched to agalsidase alfa, 18 patients of those being given a reduced-dosage regimen with agalsidase beta before swapped at different time schedules afterwards. The median duration of agalsidase alfa treatment after switching was shorter: 0.9 years (range: 0.5-1.4 years). An increase in lyso-Gb3 was seen during the shortage. All in all, the results showed no increase in the incidence of clinical events during the period of enzyme shortage19.
We have not measured Gb3 or LysoGb3 levels in plasma or in selected tissues (myocardial, renal, etc.), whether tissue deposits were reduced after sustained ERT for 24 months with agalsidase alfa in our study is not known. Nonetheless in the study of Tsuboi et al pa
Fig. 3.– Effects of agalsidase alfa treatment on Short Form-36 (SF-36) scores assessed at switch and 24 months after switching from agalsidase beta in 33 FD individuals. No statistically significant differences at 24 months compared to baseline were observed for any of the studied domains except for physical functioning. (p: 0.039).MEDICINA – Volumen 77 – Nº 3, 2017 178
tients17 experienced reduction in LV mass after 1 and 3 years of switching treatment. Also, their plasma Gb3 and lyso-Gb3 levels remained stable and showed no evidence of increasing up to 36 months after the switch. Of note for biomarkers like Gb3 the association between their plasma levels and the organ structure and function is still a matter of debate20.
Recently, Lenders et al from Germany reported the results of a retrospective analysis in adult patients with FD21. Due to the agalsidase beta shortage, 89 patients that were on agalsidase beta (1.0 mg/kg body wt) for > 1 year were nonrandomly assigned to either continue on regular-dose, receive a reduced dose of 0.3-0.5 mg/kg and a subsequent switch to 0.2 mg/kg agalsidase alfa, or directly switch to 0.2 mg/kg agalsidase alfa and were followed-up for 2 years20. All the patients after 2 years showed a stable clinical disease course with respect to serious clinical events. However, a significant decline of renal function and increase in the incidence of gastrointestinal complaints was found among patients switched. Nonetheless, albumin-to-creatinine ratio and cardiac measurements did not differ among treatment groups. The design is not a head-to-head comparison and the authors advise that comparisons between groups should be carefully interpreted. As common for all observational studies, a number of limitations are informed, including an unavoidable selection of patients. In addition, the number of patients reported was slightly lower than in a previous publication21.
Proteinuria is an important risk factor for progression of renal compromise in FD. The administration of ACEIs and/or ARBs has been demonstrated to be renoprotective in a wide variety of proteinuric renal diseases, and could consequently be vital in FD as well. Therefore, the use of such drugs is strongly recommended in FD22-24. In our cohort, antiproteinuric therapy had been started as soon as evidences of abnormal urine protein excretion were observed and this measure was maintained throughout the study period after switch. The dose of ACEIs/ARAs was kept unchanged so that no adjustment of dose was required during the 24 month-follow-up. The stabilization in urine protein excretion may be ascribed to the effect of the simultaneous use of ACEIs/ARAs plus ERT.
Two of the secondary endopoint measures in the current study were related to quality of life and body pain. The SF36 questionnaire remained basically unchanged (except for a clinically irrelevant change in the physical functioning domain) during treatment with agalsidase alfa and none of the eight domains changed more than 7 points in any patient25. Pain intensity assessed through the BPI showed a statistically significant increase in BPI in our cohort after 24 months of switching to agalsidase alfa. However, after removing from the analysis the single patient with outlier values, the statistically significant difference observed when comparing 24 months after switching vs. baseline data for physical functioning and BPI became statistically non-significant. BPI average pain item score has been used as primary measure of pain severity in several phase III, multicenter, placebo-controlled trials26 and has been profusely adopted as clinical endpoint in numerous Fabry disease studies19, 27-31. The assessment tool seems to be particularly useful for measuring patient’s outcomes in relation to the disease chronic neuropathy26. A 2-point change of the BPI average pain score and BPI severity score represents the Minimal Clinically Important Difference26 and that cuff off point has been adopted in the present study to determine improvement, impairment or stability.
We cannot rule out that multiple statistical testing in our study might lead to an increase in type I error and therefore some comparisons may have statistical significance p values purely by chance. Besides, given their subjective nature, QoL, pain and MSSI are experiences that can be influenced by patient expectation32, 33 and the tools currently available to assess them may be somewhat inaccurate and highly susceptible to bias particularly in study designs as the present.
In terms of pain intensity, we observed that it worsened in 4/33 patients. Two of these 4 patients presented with carpal tunnel syndrome confirmed by biopsy and the reported pain was specifically in hands. Pain resolved completely after carpal tunnel surgery.
The current study has several potential limitations: an uncontrolled retrospective observational nature, a heterogeneous patient population, and a relatively small number of subjects that may reduce the power to detect statistically and/or clinically relevant changes or in some cases could lead to overestimate them.
In spite of those limitations, to our knowledge, this is the first multinational study on the effects of switching agalsidase beta to agalsidase alfa for reasons other than lack of efficacy. The present study offers significant useful longitudinal information for physicians managing patients who have had their therapy switched from agalsidase beta. Our results point out that disease manifestations remain stable with agalsidase alfa. Further robust studies, adequately designed and powered, are needed to confirm if switching from agalsidase beta 1.0 mg/kg to agalsidase alfa 0.2 mg/kg does not affect rate of disease progression.
Our findings demonstrate that switching to agalsidase alfa from agalsidase beta for the treatment of FD did not significantly affect renal or cardiac functions or structure, quality of life, pain, or scoring of signs and symptoms associated with Fabry disease. Also, they confirm that the switch was largely well tolerated. Patients were able to preserve a clinically constant disease status during the switch to two year-term agalsidase alfa.
Further long-term outcome studies would be necessary to validate the findings observed in this study.
Acknowledgements: This work was supported by a grant from Shire [IIR-ARG-000137]. The funding source had no involvement in study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the article for publication. Shire has reviewed and approved the submission, but the opinions and conclusions remain those of the authors.
Conflict of interests: Francisca Masllorens and Diego Ripeau received a funding research grant from Shire Human Genetics SA to carry out the study. Francisca Masllorens has received speaker honoraria from Shire, Genzyme and Biomarin. Diego Ripeau has received speaker honoraria from Shire and Genzyme. Martin Cedrolla has received speaker honoraria from Shire and Genzyme. Luis Urtiaga has received speaker honoraria from Shire. Hernan Amartino has received speaker honoraria from Shire, Genzyme and Biomarin. Also, he received investigator’s fees from Shire and Amicus.
References
1. Brady RO, Gal AE, Bradley RM, Martensson E, Warshaw AL, Laster L. Enzymatic defect in Fabry´s disease. Ceramidetrihexosidase deficiency. N Engl J Med 1976; 276: 1163-7.
2. Desnick RJ, Ioannou YA, Eng CM. a-Galactosidase A deficiency: Fabry disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic bases of Inherited Disease, 8th ed. New York: MacGraw-Hill; 2001, p 3733-74.
3. Kolodny EH, Pastores GM. Anderson-Fabry disease: extrarenal, neurologic manifestations. J Am Soc Nephrol 2002; 13: 150-3.
4. Zarate YA, Hopkin RJ. Fabry’s disease. Lancet 2008; 372: 1427-35.
5. Branton M, Schiffmann R, Kopp JB. Natural history and treatment of renal involvement in Fabry disease. J Am Soc Nephrol 2002; 13 Suppl 2: S139-43.
6. Schiffmann R, Kopp JB, Austin HA 3rd, et al. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA 2001; 285: 2743-49.
7. Eng CM, Gurron N, Wilcox WR, et al. Safety and efficacy of recombinant human alpha-galactosidase A-replacement therapy in Fabry´s disease. N Engl J Med 2001; 345: 9-16.
8. Mehta A, Beck M, Elliott P, et al. Fabry Outcome Survey investigators. Enzyme replacement therapy with agalsidase alfa in patients with Fabry’s disease: an analysis of registry data. Lancet 2009; 374: 1986-96.
9. Banikazemi M, Bultas J, Waldek S, et al. Fabry Disease Clinical Trial Study Group. Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann Intern Med 2007; 146: 77-86.
10. Hughes DA, Elliott PM, Shah J, et al. Effects of enzyme replacement therapy on the cardiomyopathy of Anderson-Fabry disease: a randomised, double-blind, placebo-controlled clinical trial of agalsidase alfa. Heart 2008; 94: 153-8.
11. Hoffmann B. Fabry disease: recent advances in pathology, diagnosis, treatment and monitoring. Orphanet J Rare Dis 2009; 4: 21.
12. Sirrs SM, Bichet DG, Casey R, et al. Outcomes of patients treated through the Canadian Fabry disease initiative. Mol Genet Metab 2014; 111: 499-506.
13. Lin HY, Huang YH, Liao HC, et al. Clinical observations on enzyme replacement therapy in patients with Fabry disease and the switch from agalsidase beta to agalsidase alfa. J Chin Med Assoc 2014; 77: 190-7.
14. EMEA Assessment report on the shortage of Fabrazyme. Overview of Shortage Period: Spontaneous Reports from June 2009 through 15 September 2010 and Registry Data from June 2009 through 05 August 2010. In: http://www.ema.europa.eu/docs/en_GB/document_library/Other/2010/11/WC500099241.pdf; consulted 01/07/2014.
15. Levey AS, Stevens LA, Schmid CH, CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration), et al. A new equation to estimate glomerular filtration rate. Ann Intern Med 2009; 150: 604-12.
16. Politei J, Schenone AB, Cabrera G, Heguilen R, Szlago M. Fabry disease and enzyme replacement therapy in classic patients with same mutation: different formulations – different outcome? Clin Genet 2016; 89: 88-92.
17. Tsuboi K, Yamamoto H. Clinical course of patients with Fabry disease who were switched from agalsidase beta to agalsidase alfa. Genet Med 2014; 16: 766-72.
18. Pisani A, Spinelli L, Visciano B, et al. Effects of Switching from Agalsidase Beta to Agalsidase Alfa in 10 Patients with Anderson-Fabry Disease. JIMD Rep 2013; 9: 41-8.
19. Smid BE, Rombach SM, Aerts JM, et al. Consequences of a global enzyme shortage of agalsidase beta in adult Dutch Fabry patients. Orphanet J Rare Dis 2011; 6: 69.
20. Lidove O, Joly D, Barbey F, et al. Clinical results of enzyme replacement therapy in Fabry disease: a comprehensive review of literature. Int J Clin Pract 2007; 61: 293-302.
21. Lenders M, Canaan-Kühl S, Krämer J, et al. Patients with Fabry Disease after Enzyme Replacement Therapy Dose Reduction and Switch-2-Year Follow-Up. J Am Soc Nephrol 2016; 27: 952-62.
22. Ortiz A, Oliveira JP, Wanner C, Brenner BM, Waldek S, Warnock DG. Recommendations and guidelines for the diagnosis and treatment of Fabry nephropathy in adults. Nat Clin Pract Nephrol 2008; 4: 327-36.
23. Warnock DG, Daina E, Remuzzi G, West M. Enzyme replacement therapy and Fabry nephropathy. Clin J Am Soc Nephrol 2010; 5: 371-8.
24. Mehta A, West ML, Pintos-Morell G, et al. Therapeutic goals in the treatment of Fabry disease. Genet Med 2010; 12 :713-20.
25. Angst F, Aeschlimann A, Stucki G. Smallest detectable and minimal clinically important differences of rehabilitation intervention with their implications for required sample sizes using WOMAC and SF-36 quality of life measurement instruments in patients with osteoarthritis of the lower extremities. Arthritis Rheum 2001; 45: 384-91.
26. Mease PJ, Spaeth M, Clauw DJ, et al. Estimation of minimum clinically important difference for pain in fibromyalgia. Arthritis Care Res 2011; 63: 821-6.
27. Ghali J, Nicholls K, Denaro C, et al. Effect of reduced agalsidase Beta dosage in fabry patients: the Australian experience. JIMD Rep 2012; 3: 33-43.
28. Ramaswami U, Stull DE, Parini R, et al. Measuring patient experiences in Fabry disease: validation of the Fabry specific Pediatric Health and Pain Questionnaire (FPHPQ). Health Qual Life Outcomes 2012; 10: 116.
29. Whybra C, Miebach E, Mengel E, et al. A 4-year study of the efficacy and tolerability of enzyme replacement therapy with agalsidase alfa in 36 women with Fabry disease. Genet Med 2009; 11 :441-9.
30. Hoffmann B, Garcia de Lorenzo A, Mehta A, et al. Effects of enzyme replacement therapy on pain and health related quality of life in patients with Fabry disease: data from FOS (Fabry Outcome Survey). J Med Genet 2005; 42: 247-52.
31. Ries M, Mengel E, Kutschke G, et al. Use of gabapentin to reduce chronic neuropathic pain in Fabry disease. J Inherit Metab Dis 2003; 26: 413-4.
32. Turner JA, Deyo RA, Loeser JD, Von Korff M, Fordyce WE. The importance of placebo effects in pain treatment and research. JAMA 1994; 271: 1609-14.
33. Schwartz CE, Bode R, Repucci N, Becker J, Sprangers MA, Fayers PM. The clinical significance of adaptation to changing health: a meta-analysis of response shift. Qual Life Res 2006; 15: 1533-50.