MÓNICA A. COSTAS, MARÍA F. RUBIO
Laboratorio de Biología Molecular y Apoptosis, Instituto de Investigaciones Médicas Alfredo Lanari, IDIM-CONICET, Facultad de Medicina, Universidad de Buenos Aires
Resumen La autofagia es un proceso de reciclado de partes de la célula. Como se describe en esta revisión,
ocurre naturalmente preservando a las células de la acumulación de toxinas, moléculas y organelas dañadas y además permite los procesos de desarrollo y diferenciación de los tejidos. En el transcurso de la autofagia, el procesamiento de los sustratos a reciclar genera ATP, lo que constituye una fuente alternativa de energía en situaciones de estrés. En este sentido, bajo condiciones hostiles como hipoxia o falta de nutrientes, el proceso puede dispararse de modo exacerbado llevando a la muerte celular. Algunas alteraciones en su funcionamiento pueden involucrar el desarrollo de diversas patologías, tales como el daño hepático, el cáncer y las enfermedades neurodegenerativas.
Palabras clave: autofagia, mTOR, supervivencia celular
Abstract Autophagy. A strategy for cell survival. Autophagy is a process of recycling parts of the cell. As
described in this review, it occurs naturally in order to preserve cells from the accumulation of toxins, damaged molecules and organelles, and to allow processes of tissue development and differentiation. In the course of autophagy, the processing of the substrates to be recycled generates ATP, thus providing an alternative source of energy in stress situations. In this sense, under hostile conditions such as hypoxia or lack of nutrients, the autophagy process can be exacerbated leading to cell death. Some alterations in its functioning may involve the development of various pathologies, including liver damage, cancer and neurodegenerative diseases.
Key words: autophagy, mTOR, cell survival
Recibido: 26-V-2017 Aceptado: 5-VII-2017
Dirección postal: Dra. Mónica A. Costas, Laboratorio de Biología Molecular y Apoptosis, Instituto de Investigaciones Médicas Alfredo Lanari, IDIM-CONICET, Facultad de Medicina, Universidad de Buenos Aires, Combatientes de Malvinas 3150, 1427 Buenos Aires, Argentina
e-mail: mcostasra@hotmail.com
Los organismos subsisten y se preservan naturalmente combatiendo patógenos y enfermedades a través de sistemas que funcionan en equilibrio homeostático. Tal es el caso del sistema inmunológico y el endocrino, que utilizan células especializadas y mediadores químicos como los anticuerpos y las hormonas.
El individuo debe enfrentar permanentemente daños a sus células y tejidos debido no solo a señales del entorno sino también al deterioro propio de la madurez y el envejecimiento. Así es que el organismo posee mecanismos para protegerse de las células dañadas, ya sea deshaciéndose de ellas o bien impidiendo que se propaguen, de modo de evitar una descendencia de células portadoras de errores y mutaciones que podrían derivar en estados patológicos.
Entre los mecanismos que suprimen la existencia de células dañadas, perjudiciales para la salud o innecesarias para el correcto funcionamiento de los aparatos y sistemas, se encuentra la “apoptosis”, un proceso de muerte celular altamente controlado en el cual esa célula se sacrifica para preservar al tejido, órgano o individuo. Es un tipo de muerte celular en el cual la membrana plasmática celular se preserva hasta la última etapa del proceso, luego de la fragmentación del ADN y la pérdida de núcleo, evitando la fuerte reacción inflamatoria ocasionada por el derrame del contenido celular en el microentorno tisular1, 2.
Otro mecanismo, la “senescencia”, evita la propagación de mutaciones acumuladas en las células en los sucesivos ciclos de replicación debido ya sea a señales tóxicas o al propio acortamiento telomérico que ocurre en cada ciclo celular3-6. Además, las células no pueden replicarse indefinidamente. Más allá de los daños y las mutaciones acumulados en la vida, el mencionado acortamiento de los telómeros causa errores de replicación comprometiendo material genético que codifica para funciones vitales. En la senescencia, las células no mueren, solo se detiene su ciclo de división celular de modo irreversible, evitando dejar progenie con errores7. Si bien siguen activas metabólicamente, cambian su patrón de expresión génica produciendo otros factores y proteínas diferentes a las que producen en su estado nativo original8.
Autofagia en el reciclado de organelas y moléculas dañadas
Además de los dos procesos mencionados por los cuales una célula sufre apoptosis o entra en senescencia, existe un proceso alternativo de supervivencia celular en el cual sacrifica partes o moléculas dañadas para preservar su vida. Este proceso se conoce como “autofagia”9-13 y fue descubierto por el científico japonés Yoshinori Ohsumi, galardonado con el Premio Nobel de Medicina 2016 por estos hallazgos. La importancia de su descubrimiento ha adquirido en los últimos años la misma relevancia que los procesos de apoptosis y senescencia, dada la participación de la autofagia en los complejos procesos que controlan la vida celular y en el desarrollo de numerosas patologías. A pesar de que la supervivencia celular podría ser considerada su primer objetivo, no siempre ocurre así, pues cuando la autofagia se manifiesta de modo exacerbado puede llevar a la muerte celular14-16.
Existen dos tipos de autofagia: la macroautofagia en la cual se degradan organelas dañadas o disfuncionales, y la microautofagia en la cual se degradan moléculas dañadas, truncas o mal procesadas que no adquieren la conformación adecuada17, 18.
La macroautofagia resulta fundamental en la eliminación de mitocondrias (mitofagia) disfuncionales y el mantenimiento del número adecuado de las mismas según el tipo de tejido y su función17, 19. La mitofagia es un proceso importante en el desarrollo de tumores15, 17, 20, 21 y enfermedades neurodegenerativas como la enfermedad de Alzheimer, donde se acumulan mitocondrias disfuncionales por fallas en dicho proceso19.
La microautofagia desempeña un rol importante en la detoxificación de los tejidos y su alteración permite la acumulación de proteínas mal plegadas y toxinas que contribuyen al desarrollo de varias enfermedades que involucran la muerte celular. Este es el caso del epitelio pulmonar de pacientes con fibrosis quística, donde los acúmulos de moléculas no degradadas por la disfunción de la autofagia forman agregosomas22.
De acuerdo a trabajos muy recientes, existen evidencias de la importancia de la autofagia para el mantenimiento del equilibrio homeostático de proteínas a nivel presináptico que preserva el correcto funcionamiento de la conducción nerviosa23. En este proceso, más allá de la macro y la microautofagia mencionadas más arriba, el proceso involucra un tercer mecanismo de autofagia mediado por chaperonas.
Otras evidencias muestran que en la corteza cerebral de pacientes con esquizofrenia existe una expresión disminuida de BECN1, gen que codifica para la proteína Beclin1 requerida para el desarrollo del proceso autofágico. Además, la inducción de autofagia es parte del mecanismo de acción del litio (usualmente incluido en la medicación de trastornos bipolares), resaltando su rol en el desarrollo y el tratamiento de desórdenes psiquiátricos24.
La autofagia también participa en el desarrollo de cardiomiopatías. Los cardiomiocitos la requieren para evitar la acumulación de proteínas mal plegadas y organelas dañadas25. Si bien en condiciones basales ocurre a muy baja escala, este proceso aumenta ante situaciones de estrés y en condiciones crónicas de arritmias, isquemia e hipertrofia25.
Autofagia y diferenciación tisular
La autofagia es elemental para la renovación celular y forma parte del proceso de desarrollo y de la diferenciación celular en condiciones fisiológicas normales. Participa en los procesos de diferenciación de las células maduras que forman parte de cada tejido sano a partir de células madre indiferenciadas, así como en los de regeneración tisular y de transformación adipocítica26, 27. Precisamente, la diferenciación celular implica el reciclado de parte de sus moléculas y organelas para dar lugar a una nueva célula con morfología y función específicas, como se ha demostrado en la maduración renal28 y también en la diferenciación a osteoblastos a partir de células pluripotentes inducidas por reprogramación genética29. Más aún, Nanog (uno de los marcadores de fenotipo de “célula madre” que se utiliza junto a otros tres para reprogramar células maduras diferenciadas a células pluripotentes30) induce autofagia en condiciones de hipoxia31.
El síndrome metabólico se caracteriza por insulino-resistencia, obesidad y una reacción inflamatoria generalizada. De acuerdo a estudios en modelos equinos de la enfermedad, se ha demostrado que la autofagia evita la diferenciación a condrocitos de las células madre residentes en el tejido adiposo32.
En cuanto a la maduración de células hematopoyéticas, también se ha demostrado que la autofagia juega un rol importante en el mantenimiento del balance entre la auto-renovación de las células madre y su división asimétrica para generar células diferenciadas33.
Autofagia y metabolismo energético
Cuando se produce la autofagia, además de eliminarse partes de la célula que no sirven o que resultan perjudiciales para su sana supervivencia, los desechos se aprovechan como metabolitos para producir energía. Por esta razón, la señal de inicio de autofagia no necesariamente es provista por daño intracelular15, 34-36. En condiciones de estrés por falta de nutrientes u oxígeno, algunas células disparan el proceso de autofagia sacrificando parte de sí mismas para producir energía y sobrevivir en condiciones adversas37, 38.
Más allá de la supervivencia celular específica, la autofagia es necesaria para la supervivencia de los mamíferos. Mediante experimentos realizados en ratones en condiciones de ayuno, se demostró que si se eliminaba Atg7 (Autophagy related gene), un gen esencial para el desarrollo de autofagia, los animales se morían a poco de nacer35. Además, se observó que si se los alimentaba, la supervivencia se prolongaba solo por un corto período, demostrando que el proceso era esencial para la supervivencia, independientemente del ayuno35. Si bien los animales adultos poseen reservas para subsistir en ayuno, en ellos la deleción del mismo gen también resulta letal. Tanto la degradación de glucógeno para producir glucosa, como el catabolismo lipídico para la producción de glicerol y ácidos grasos son ineficientes si las células tienen el defecto metabólico de no poder realizar autofagia. Las reservas se agotan rápidamente en un catabolismo acelerado y, al no poder mantener los niveles circulantes de glucosa, los animales mueren rápidamente por hipoglucemia, daño hepático y neurodegeneración35. Por lo tanto, la autofagia, además de una respuesta al estrés, es un proceso requerido normalmente para mantener la homeostasis en el metabolismo.
Autofagia y cáncer
La autofagia resulta clave en el control del desarrollo tumoral y puede desempeñar un rol dual, favoreciendo tanto el crecimiento como la eliminación de las células cancerígenas16, 20, 21, 39. El rol pro-tumoral se evidencia cuando los tumores en crecimiento adquieren un tamaño considerable, dificultando la llegada de nutrientes y oxígeno al interior de la masa tumoral mientras no se desarrolle el proceso angiogénico. En esta etapa, la autofagia constituye una estrategia de supervivencia hasta que se logra una buena irrigación sanguínea y la consiguiente provisión de nutrientes y oxígeno20. Por el contrario, es sabido que la inducción exacerbada de autofagia a través de drogas quimioterapéuticas puede llevar a la muerte celular, mostrando así un rol antitumoral40. Sin embargo, existen evidencias de que las células tumorales tienen, en condiciones basales, mayores niveles de autofagia que las células sanas normales y esto podría contribuir a una mayor resistencia a estos tratamientos21, 41. También se ha descripto que, dentro de la heterogeneidad de la población celular de un tumor, las células madre tumorales serían las de mayores niveles de autofagia42, imprescindible para su autopreservación debido a que se desarrollan en nichos hipóxicos43, 44.
En esta línea de razonamiento, una autofagia basal sería necesaria para la tumorigénesis, así como para la propagación en los focos metastásicos. De hecho, las células madre tumorales serían las responsables de este proceso, pudiendo iniciar el crecimiento de tumores secundarios luego de la transición epitelio-mesenquimática, migración e invasión hacia los focos metastásicos42, 43, 45. En la transición epitelio-mesenquimática, algunas células tumorales sufren un proceso de desdiferenciación desde un fenotipo epitelial hacia uno mesenquimático a través de rearreglos del citoesqueleto y cambios en el patrón de expresión génica, donde se inhibe la expresión de E-cadherinas, responsables de los contactos célula-célula, y aumenta la expresión de vimentina43, 46. Una vez que las células migratorias llegan a destino, este proceso se revierte47, con la consiguiente reconstrucción del tumor. Para ello, es necesario que la población de células migratorias incluya células madre tumorales45, 48.
Actualmente existen numerosas moléculas marcadoras (CD133, CD44, SOX2, Nanog, Oct 4 entre otras30, 49-51) y procedimientos experimentales que permiten identificar poblaciones de células madre tumorales. Tal es el caso del crecimiento clonogénico, la generación de tumoresferas46, 52 y la retención disminuida de fluorocromos53 debido a una mayor expresión de receptores MDR (transportadores ABG con actividad ATPasa) que lo bombean al espacio extracelular, de modo similar a lo que ocurre con las drogas quimioterapéuticas54. La expresión de genes de pluripotencia SOX2, Nanog y Oct4, así como la habilidad de generar tumoresferas, son comunes a las células madre normales42. A su vez, estas características también son compartidas con las células mesenquimales, por lo que la identificación de estas subpoblaciones aún no está totalmente resuelta, con excepción de la capacidad que tienen las células madre de regenerar el tumor cuando se las implanta en modelos animales42, 44, 45. A pesar de tantas similitudes entre las distintas poblaciones celulares, estudios con inhibidores específicos de la autofagia demostraron que las células madre tumorales pierden su viabilidad, pero no las mesenquimales43, 44, lo que resalta la importancia de la preservación del proceso de autofagia como una propiedad selectiva entre ambas poblaciones43.
En resumen, la autofagia desempeña distintos roles en el desarrollo tumoral: por un lado tiene un rol antitumoral si se induce de modo exacerbado, y por el otro previene la muerte de células tumorales una vez iniciada la tumorigénesis, previa inducción de angiogénesis. De acuerdo con esto, resulta un proceso atractivo como blanco terapéutico o un mecanismo de preservación de las células tumorales, dependiendo de la etapa de la enfermedad.
El proceso de autofagia
Entre los principales eventos que una célula detecta para aumentar los niveles de autofagia se encuentran el mal plegamiento de proteínas, organelas dañadas que funcionan de modo anómalo o que aumentan su número de modo inadecuado, y una carencia de nutrientes o hipoxia.
La vía de señalización mTOR (mammalian target of rapamycin) consta de dos ramas principales, cada una mediada por un mTOR. La vía rapamicina-sensible mTOR1 controla varios caminos que, colectivamente, determinan la masa o tamaño de la célula. La vía rapamicina-insensible mTOR2 controla la actina del citoesqueleto y, por lo tanto, determina la forma de la célula. La vía mTOR1 es una de las más importantes en el control de la autofagia, tiene un rol fundamental censando carencia de nutrientes, hipoxia y balance metábólico55, 56, tal como se muestra en la Fig. 1.
El proceso de autofagia puede dividirse en fases: 1) nucleación, 2) elongación, 3) formación del autofagosoma maduro, 4) fusión, 5) degradación, y 6) reciclaje (Fig. 2). La fase de nucleación está regulada por el complejo mTORC1, un complejo formado por 5 proteínas: mTOR, Raptor (regulatory-associated protein of mTOR), mLST8 (proteína letal 8 de mamíferos con SEC13), PRAS 40 (substrato de 40 kDa de Akt rico en prolina) y DEPTOR, cuya regulación depende de la relación AMP/ATP la cual, a su vez, está relacionada con el estado nutricional y metabólico (Fig. 1). Este complejo y las proteínas río abajo se regulan por fosforilación/desfosforilación y activan las proteínas encargadas de la iniciación del proceso, vía activación de UKL1/UKL2 (proteínas quinasa tipo uridina 1 y 2, respectivamente) (Fig. 2). Gran parte del proceso es mediado por las proteínas de la familia ATG (proteínas iniciadoras de autofagia), las cuales se unen a las proteínas u organelas dañadas, marcándolas para la formación de fagóforos (vesículas de doble membrana que contienen el material a reciclar). Algunos miembros de la familia se unen a los fagóforos, promoviendo su maduración y fusión con el lisosoma para formar el autofagosoma18. Esta fusión permite que las enzimas lisosomales se ocupen de la degradación enzimática de los sustratos, cuyos productos (aminoácidos, lípidos) son luego exportados al citoplasma para su reutilización (Fig. 2).
Más precisamente, bajo las señales de estrés mencionadas, se activan una serie de proteínas reguladoras de la autofagia como mTORC1, además de otras quinasas como AMPK (proteína quinasa dependiente de adenosina monofosfato) y PKA (proteína quinasa A) (Fig. 1) que, a su vez, regulan la actividad del complejo quinasa-quinasa ULK, compuesto por ULK1/2, FIP200 y otras ATGs (Fig. 2). En la iniciación del proceso, primero la quinasa ULK1 activa por fosforilación a la proteína Beclin1 (también conocida como ATG6) que forma parte del complejo iniciador de la nucleación del fagóforo. Posteriormente, se
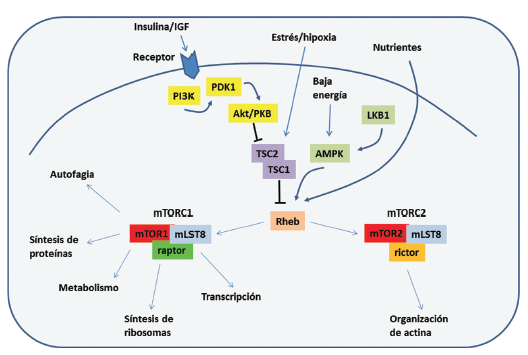
Fig. 1.– La red de señalización mTOR consta de dos ramas principales, cada una mediada por un mTORC. La vía rapamicina sensible mTORC1 controla varios caminos que colectivamente determinan la masa o tamaño de la célula. mTORC1 y mTORC2 responden a factores de crecimiento (insulina / IGF), al estado energético de las células, nutrientes (aminoácidos) y a estrés. mTORC1 y mTORC2 son multiméricos, aunque se esquematizan como monómeros. Las flechas representan la activación, mientras que las barras representan la inhibición. PI3K (fosfatidilinositol 3 quinasa), Akt/PKB (proteína quinasa B), PDK (proteína quinasa 1-dependiente de fosfatidilinositol), TSC (tuberous sclerosis complex) 1 y 2, LKB1 (serina treonina quinasa codificada por el gen LKB1), AMPK (quinasa dependiente de adenosina monofoasfato), Rheb (RAS homologue enriched in brain) es una GTPasa, mLST8 (proteína letal 8 de mamíferos con SEC13), RAPTOR (regulatory-associated protein of mTOR), RICTOR (rapamycin-insensitive companion of mammalian target of rapamycin)
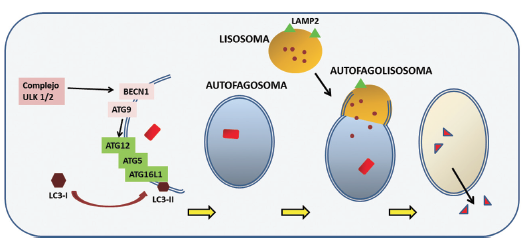
Fig. 2.– Génesis y desarrollo del autofagosoma y participación de las diferentes proteínas ATG en sus 6 etapas. Se inicia por activación de complejo quinasa-quinasa ULK1/2 (proteínas quinasa tipo uridina 1 y 2, respectivamente), luego ocurre la formación del fagóforo o nucleación y posteriormente la conjugación del mismo con el complejo ATG5/ATG12/ATG16 (proteínas iniciadoras de autofagia 5, 12 y 16). Continúa con el procesamiento de LC3 (microtubule-associated protein light chain 3) y la inserción en la membrana del fagóforo. Así se produce la formación del autofagosoma que luego se fusiona con el lisosoma (autofagolisosoma) adonde ocurre la degradación del substrato cuyos productos son liberados al citoplasma.
genera PIP3 (3-fosfatidil inositol fosfato), esencial para el reclutamiento de otras proteínas ATGs sobre las dobles membranas que provienen del retículo endoplásmico, el complejo de Golgi y las mitocondrias57, 58. La expansión de la doble membrana lipídica para la posterior formación del autofagosoma se produce por la interacción del fagóforo con el complejo proteico ATG5/ATG12. La conjugación de este dímero requiere la acción enzimática de ATG7 y ATG1059. Finalmente, la unión de ATG16 al complejo ATG5/ATG12 produce su multimerización y formación del complejo que permite la inserción de la forma II de LC3 (microtubule-associated protein light chain 3, también conocida como ATG8) en la membrana del fagóforo37. La proteína LC3 se sinteriza como pro-LC3, la cual es seccionada por ATG4, generando la forma activa LC3-I en el citosol. Posteriormente, al LC3-I se incorporan lípidos por la acción de ATG7 y ATG3 que catalizan su unión a los residuos de fosfatidiletanolamina presentes en la membrana del fagóforo60. La unión de LC3-II al fagóforo se requiere para el cierre de la vesícula luego de la captación de los sustratos a procesar y así dar origen al autofagosoma61. Una vez formado, el complejo ATG5/ATG12/ATG16 y el LC3-II37se libera para formar el autofagosoma desnudo. Cuando éste se fusiona con los lisosomas por acción de Rab7, Lamp1 y Lamp2, se forma el autofagolisosoma37. Finalmente, los substratos se degradan por la acción de las enzimas lisosomales y los productos de la reacción se liberan al citoplasma para su futura reutilización61 (Fig. 2).
Diversos fármacos pueden intervenir en la regulación de la autofagia; puede ser inhibida por 3-MA (3 metil-adenina), por inhibidores del complejo PI3K clase III (fosfatidil inositol 3-quinasa III) y por spautin-1, que promueve la degradación de los complejos PI3K de clase III36, 62. Una vez que se inicia la formación de fagóforos, compuestos tales como la verteporfina pueden interferir con los motivos de la región que interactúan con LC3 y bloquear el reclutamiento selectivo de cargas como las mitocondrias63. El tráfico de autofagosomas al lisosoma es facilitado por el citoesqueleto. Por lo tanto, la desestabilización de los microtúbulos por los alcaloides de la vinca puede bloquear la maduración de los autofagosomas, mientras que la estabilización por el taxol puede aumentar la fusión entre las vacuolas autofágicas y los lisosomas64. Los agentes lisosomotrópicos que aumentan el pH lisosómico, como cloroquina, NH4Cl y monensina, interfieren con la función lisosómica y bloquean la autofagia en una etapa tardía62, 66. El paso final de la vía de autofagia también puede ser bloqueado por inhibidores de enzimas lisosómicas, tales como E64d, pepstatina A, y bafilomicina A162.
La rapamicina es un inhibidor de la vía mTORC1 que interacciona con este complejo a través de la unión con la inmunofilina FKBP12. En trabajos previos de nuestro grupo hemos demostrado el rol de esta droga en la inducción de autofagia36. Por otro lado, también hemos observado que la expresión en altos niveles de RAC3, un coactivador de receptores nucleares usualmente sobreexpresado en varios tipos tumorales67-71, tiene un rol protector contra la autofagia, probablemente preservando tempranamente a la célula de la muerte por autofagia en la iniciación tumoral, acorde al rol dual en tumorigénesis discutido más arriba. Sin embargo, es interesante destacar que la hipoxia inhibe la expresión de RAC3, generando así un ambiente permisivo para el proceso de autofagia que preserva la supervivencia de las células tumorales ante la carencia de oxígeno y nutrientes hasta que ocurre la angiogénesis36.
Conclusiones y direcciones futuras
En la mayoría de las células, la autofagia se produce a niveles basales bajos, pero a menudo bajo condiciones como el estrés, aumenta para mantener la supervivencia celular, como una respuesta citoprotectora esencial. De acuerdo con lo expuesto en esta revisión, las alteraciones del proceso autofágico están implicadas en la fisiopatología de cardiomiopatías, enfermedades infecciosas, enfermedad de Crohn y trastornos neurodegenerativos19, 38. También se ha sugerido que la sobreactivación de la autofagia promueve la supervivencia de células cancerígenas en el microambiente tumoral in vivo y contribuye a la resistencia a quimioterapias y estrés, promoviendo la metástasis y la latencia62.
Por lo tanto, aunque la rapamicina y otros inhibidores de mTOR han sido probados como terapéuticos anticancerosos, evidencia creciente sugiere que tanto la activación como la inhibición de la autofagia pueden promover efectos aún no esclarecidos, induciendo tanto la supervivencia como la muerte de las células cancerosas39, pero afectando además otras respuestas biológicas.
Dado que los defectos en las vías autofágicas han sido implicados en diversos trastornos, desde enfermedades inflamatorias a cánceres y neurodegeneración, el estímulo o la inhibición de la autofagia podrían ser utilizados para su tratamiento. Con el avance en nuestra comprensión de las vías de autofagia, el diseño de moduladores de autofagia proporcionará herramientas para el desarrollo de nuevas terapias para varias enfermedades.
Conflicto de intereses: Ninguno para declarar
Bibliografía
1. Costas MA. Vida y muerte de la célula: las señales intracelulares. Medicina (B Aires) 2006; 66: 281-4.
2. Colo GP, Rubio MF, Nojek IM, et al. The p160 nuclear receptor co-activator RAC3 exerts an anti-apoptotic role through a cytoplasmatic action. Oncogene 2008; 27: 2430-44.
3. Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 2005; 120: 513-22.
4. Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell 2007; 130: 223-33.
5. Vicencio JM, Galluzzi L, Tajeddine N, et al. Senescence, apoptosis or autophagy? When a damaged cell must decide its path–a mini-review. Gerontology 2008; 54: 92-9.
6. Fernandez Larrosa PN, Ruiz Grecco M, Mengual Gomez D, et al. RAC3 more than a nuclear receptor coactivator: a key inhibitor of senescence that is downregulated in aging. Cell Death Dis 2015; 6: e1902.
7. Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 2007; 8: 729-40.
8. Davalos AR, Coppe JP, Campisi J, Desprez PY. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev 2010; 29: 273-83.
9. Takeshige K, Baba M, Tsuboi S, Noda T, Ohsumi Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J Cell Biol 1992; 119: 301-11.
10. Baba M, Osumi M, Scott SV, Klionsky DJ, Ohsumi Y. Two distinct pathways for targeting proteins from the cytoplasm to the vacuole/lysosome. J Cell Biol 1997; 139: 1687-95.
11. Mizushima N, Noda T, Yoshimori T, et al. A protein conjugation system essential for autophagy. Nature 1998; 395: 395-8.
12. Yamamoto H, Fujioka Y, Suzuki SW, et al. The intrinsically disordered protein Atg13 mediates supramolecular assembly of autophagy initiation complexes. Dev Cell 2016; 38: 86-99.
13. Ohsumi Y. Historical landmarks of autophagy research. Cell Res 2014; 24: 9-23.
14. Simone C. Signal-dependent control of autophagy and cell death in colorectal cancer cell: the role of the p38 pathway. Autophagy 2007; 3: 468-71.
15. Jacob JA, Salmani JM, Jiang Z, et al. Autophagy: An overview and its roles in cancer and obesity. Clin Chim Acta 2017; 468: 85-9.
16. Galluzzi L, Vicencio JM, Kepp O, et al. To die or not to die: that is the autophagic question. Curr Mol Med 2008; 8: 78-91.
17. Kulikov AV, Luchkina EA, Gogvadze V, Zhivotovsky B. Mitophagy: Link to cancer development and therapy. Biochem Biophys Res Commun 2017; 482: 432-9.
18. Reggiori F, Ungermann C. Autophagosome maturation and fusion. J Mol Biol 2017; 429: 486-96.
19. Kerr JS, Adriaanse BA, Greig NH, et al. Mitophagy and Alzheimer’s disease: cellular and molecular mechanisms. Trends Neurosci 2017; 40: 151-66.
20. Jin S, White E. Role of autophagy in cancer: management of metabolic stress. Autophagy 2007; 3: 28-31.
21. Tsuchihara K, Fujii S, Esumi H. Autophagy and cancer: dynamism of the metabolism of tumor cells and tissues. Cancer Lett 2009; 278: 130-8.
22. Luciani A, Villella VR, Esposito S, et al. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat Cell Biol 2010; 12: 863-75.
23. Wang YC, Lauwers E, Verstreken P. Presynaptic protein homeostasis and neuronal function. Curr Opin Genet Dev 2017; 44: 38-46.
24. Schneider JL, Miller AM, Woesner ME. Autophagy and schizophrenia: a closer look at how dysregulation of neuronal cell homeostasis influences the pathogenesis of schizophrenia. Einstein J Biol Med 2016; 31: 34-9.
25. Hashemzaei M, Entezari Heravi R, Rezaee R, Roohbakhsh A, Karimi G. Regulation of autophagy by some natural products as a potential therapeutic strategy for cardiovascular disorders. Eur J Pharmacol 2017; 802: 44-51.
26. Guo L, Huang JX, Liu Y, et al. Transactivation of Atg4b by C/EBPbeta promotes autophagy to facilitate adipogenesis. Mol Cell Biol 2013; 33: 3180-90.
27. Baerga R, Zhang Y, Chen PH, Goldman S, Jin S. Targeted deletion of autophagy-related 5 (atg5) impairs adipogenesis in a cellular model and in mice. Autophagy 2009; 5: 1118-30.
28. Zhang C, Li W, Wen J, Yang Z. Autophagy is involved in mouse kidney development and podocyte differentiation regulated by Notch signalling. J Cell Mol Med 2017; doi: 10.1111/jcmm.13061. [Epub ahead of print]
29. Ozeki N, Hase N, Hiyama T, et al. MicroRNA-211 and autophagy-related gene 14 signaling regulate osteoblast-like cell differentiation of human induced pluripotent stem cells. Exp Cell Res 2017.
30. Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature 2007; 448: 313-7.
31. Hasmim M, Janji B, Khaled M, et al. Cutting Edge: nanog activates autophagy under hypoxic stress by binding to BNIP3L promoter. J Immunol 2017; 198: 1423-8.
32. Marycz K, Kornicka K, Grzesiak J, Smieszek A, Szlapka J. Macroautophagy and selective mitophagy ameliorate chondrogenic differentiation potential in adipose stem cells of equine metabolic syndrome: new findings in the field of progenitor cells differentiation. Oxid Med Cell Longev 2016; 2016: 3718468.
33. Riffelmacher T, Simon AK. Mechanistic roles of autophagy in hematopoietic differentiation. FEBS J 2016; 284: 1008-20.
34. Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol 2002; 192: 1-15.
35. Guo JY, White E. Autophagy, metabolism, and cancer. Cold Spring Harb Symp Quant Biol 2016; 81: 73-8.
36. Fernandez Larrosa PN, Alvarado CV, Rubio MF, et al. Nuclear receptor coactivator RAC3 inhibits autophagy. Cancer Sci 2012; 103: 2064-71.
37. Kaizuka T, Morishita H, Hama Y, et al. An autophagic flux probe that releases an internal control. Mol Cell 2016; 64: 835-49.
38. Ravikumar B, Vacher C, Berger Z, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 2004; 36: 585-95.
39. Hippert MM, O’Toole PS, Thorburn A. Autophagy in cancer: good, bad, or both? Cancer Res 2006; 66: 9349-51.
40. Liu JJ, Lin M, Yu JY, Liu B, Bao JK. Targeting apoptotic and autophagic pathways for cancer therapeutics. Cancer Lett 2011; 300: 105-14.
41. Song J, Qu Z, Guo X, et al. Hypoxia-induced autophagy contributes to the chemoresistance of hepatocellular carcinoma cells. Autophagy 2009; 5: 1131-44.
42. Jordan CT, Guzman ML, Noble M. Cancer stem cells. N Engl J Med 2006; 355: 1253-61.
43. Marcucci F, Ghezzi P, Rumio C. The role of autophagy in the cross-talk between epithelial-mesenchymal transitioned tumor cells and cancer stem-like cells. Mol Cancer 2017; 16: 3.
44. Lei Y, Zhang D, Yu J, et al. Targeting autophagy in cancer stem cells as an anticancer therapy. Cancer Lett 2017; 393: 33-9.
45. Soltysova A, Altanerova V, Altaner C. Cancer stem cells. Neoplasma 2005; 52: 5.
46. Anwar TE, Kleer CG. Tissue-based identification of stem cells and epithelial-to-mesenchymal transition in breast cancer. Hum Pathol 2013; 44: 1457-64.
47. Bedi U, Mishra VK, Wasilewski D, Scheel C, Johnsen SA. Epigenetic plasticity: a central regulator of epithelial-to-mesenchymal transition in cancer. Oncotarget 2014; 5: 2016-29.
48. Tysnes BB, Bjerkvig R. Cancer initiation and progression: involvement of stem cells and the microenvironment. Biochim Biophys Acta 2007; 1775: 283-97.
49. Draffin JE, McFarlane S, Hill A, Johnston PG, Waugh DJ. CD44 potentiates the adherence of metastatic prostate and breast cancer cells to bone marrow endothelial cells. Cancer Res 2004; 64: 5702-11.
50. Williams K, Motiani K, Giridhar PV, Kasper S. CD44 integrates signaling in normal stem cell, cancer stem cell and (pre)metastatic niches. Exp Biol Med (Maywood) 2013; 238: 324-38.
51. Li Z. CD133: a stem cell biomarker and beyond. Exp Hematol Oncol 2013; 2: 17.
52. Tirino V, Desiderio V, Paino F, Papaccio G, De Rosa M. Methods for cancer stem cell detection and isolation. Methods Mol Biol 2012; 879: 513-29.
53. Seigel GM, Campbell LM. High-throughput microtiter assay for Hoechst 33342 dye uptake. Cytotechnology 2004; 45: 155-60.
54. Januchowski R, Wojtowicz K, Andrzejewska M, Zabel M. Expression of MDR1 and MDR3 gene products in paclitaxel-, doxorubicin- and vincristine-resistant cell lines. Biomed Pharmacother 2014; 68: 111-7.
55. Cam H, Easton JB, High A, Houghton PJ. mTORC1 signaling under hypoxic conditions is controlled by ATM-dependent phosphorylation of HIF-1alpha. Mol Cell 2010; 40: 509-20.
56. Easton JB, Houghton PJ. mTOR and cancer therapy. Oncogene 2006; 25: 6436-46.
57. Mauro-Lizcano M, Esteban-Martinez L, Seco E, et al. New method to assess mitophagy flux by flow cytometry. Autophagy 2015; 11: 833-43.
58. Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140: 313-26.
59. Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007; 3: 452-60.
60. Rekas A, Alattia JR, Nagai T, Miyawaki A, Ikura M. Crystal structure of venus, a yellow fluorescent protein with improved maturation and reduced environmental sensitivity. J Biol Chem 2002; 277: 50573-8.
61. Zhou C, Zhong W, Zhou J, et al. Monitoring autophagic flux by an improved tandem fluorescent-tagged LC3 (mTagRFP-mWasabi-LC3) reveals that high-dose rapamycin impairs autophagic flux in cancer cells. Autophagy 2012; 8: 1215-26.
62. Vakifahmetoglu-Norberg H, Xia HG, Yuan J. Pharmacologic agents targeting autophagy. J Clin Invest 2015; 125: 5-13.
63. Gomez VE, Giovannetti E, Peters GJ. Unraveling the complexity of autophagy: potential therapeutic applications in pancreatic ductal adenocarcinoma. Semin Cancer Biol 2015; 35: 11-9.
64. Hua F, Shang S, Hu ZW. Seeking new anti-cancer agents from autophagy-regulating natural products. J Asian Nat Prod Res 2017; 19: 305-13.
65. Datta S, Choudhury D, Das A, et al. Paclitaxel resistance development is associated with biphasic changes in reactive oxygen species, mitochondrial membrane potential and autophagy with elevated energy production capacity in lung cancer cells: A chronological study. Tumour Biol 2017; 39: 1010428317694314.
66. Kuzu OF, Toprak M, Noory MA, Robertson GP. Effect of lysosomotropic molecules on cellular homeostasis. Pharmacol Res 2017; 117: 177-84.
67. Anzick S, Kononen J, Walker R, et al. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science 1997; 277: 965-8.
68. Colo GP, Rosato RR, Grant S, Costas MA. RAC3 down-regulation sensitizes human chronic myeloid leukemia cells to TRAIL-induced apoptosis. FEBS Lett 2007; 581: 5075-81.
69. Gnanapragasam VJ, Leung HY, Pulimood AS, Neal DE, Robson CN. Expression of RAC 3, a steroid hormone receptor co-activator in prostate cancer. Br J Cancer 2001; 85: 1928-36.
70. Ma G, Ren Y, Wang K, He J. SRC-3 has a role in cancer other than as a nuclear receptor coactivator. Int J Biol Sci 2011; 7: 664-72.
71. Sakakura C, Hagiwara A, Yasuoka R, et al. Amplification and over-expression of the AIB1 nuclear receptor co-activator gene in primary gastric cancers. Int J Cancer 2000; 89: 217-23.