ELOISA MALBRÁN ¹, ALEJANDRA MENÉNDEZ ², ALEJANDRO MALBRÁN ¹
¹ Unidad de Alergia, Asma e Inmunología Clínica, ² Asociación Argentina de Pacientes con Angioedema Hereditario,
Buenos Aires, Argentina
Abstract The benefits of the worldwide approval of new drugs for the treatment of acute C1-INH-HAE attacks
may still not reach all patients. Identifying the current barriers in the access to medication, as well as conducting a detailed assessment of the progress in this area, is essential to achieve universal treatment. Two hundred and twenty five patients registered in the Argentina Hereditary Angioedema Patient Association (AHAEPA) were randomly selected and invited to participate in a web based questionnaire on accessibility to icatibant and pdC1-INH, self-treatment, delay to treatment, and coverage. The data retrieved was compared to our previous reports in 2008 and 2013. We collected 156/225 answers. One hundred and eighteen (76%) patients have either pdC1-INH (n = 86), icatibant (n = 10) or both (n = 22), while 38 (24%) do not have access to treatment. In 2008, 26% had access while 82% had it in 2013. Thirty-two subjects (22%) self-inject themselves, similar to 29% in 2013, even though between studies, widespread self-injection training activities have taken place. However, considering injections by proxy, home treatment reached 56%. Only half of the patients decide to receive treatment early during the attack. Ninety-nine patients (63%) have full coverage, thirty (19%) have no coverage at all and the rest only obtain partial reimbursement. Twenty-nine families (31%) share a single treatment dose of the medication, better than 36% in 2013. Argentina’s C1-INH-HAE patients had a sustained improvement in their access to medication. Efforts should continue to further improve accessibility and optimal management of HAE acute attacks to all patients in the country.
Key words: angioedema, hereditary angioedema, C1 inhibitor, access
Resumen Tratamiento del ataque agudo de angioedema hereditario por deficiencia del inhibidor de C1
en Argentina. La aprobación mundial de los medicamentos para el ataque agudo del angioedema hereditario (HAE) no beneficia a todos los pacientes. Es necesario conocer las barreras de acceso a la medicación para el tratamiento universal. Doscientos veinticinco pacientes, registrados en la Asociación de Pacientes con Angioedema Hereditario (AHAEPA), fueron encuestados por internet acerca de su accesibilidad al icatibant y al concentrado del inhibidor de C1 (pdC1-INH), a la auto inyección de la medicación, al retraso del tratamiento y a la cobertura del medicamento. Comparamos esta información con la obtenida en nuestros estudios de 2008 y 2013. Recolectamos 156/225 respuestas. Ciento dieciocho (76%) pacientes tienen pdC1-INH (n = 86), icatibant (n = 10) o ambos (n = 22), mientras que 38 (24%) no tienen medicación. En 2008, 26% tenían acceso y en 2013, 82%. Treinta y dos (22%) se autoinyectan la medicación, similar al 29% en 2013. Sumando las aplicaciones por profesionales de la salud o familiares en la casa, el tratamiento fuera de las instituciones médicas alcanza el 56%. Solo la mitad decide tratarse tempranamente. Noventa y nueve (63%) tiene cobertura del 100%, 30 (19%) no tiene ningún tipo de cobertura, y el resto la tiene en forma parcial. Veintinueve familias (31%), solo tienen una dosis de tratamiento para todos, mejor que el 36% en 2013. Los pacientes con C1-INH-HAE han tenido una mejoría sustancial en el acceso a la medicación. Igualmente, los esfuerzos deben continuar para mejorar la accesibilidad y tratamiento óptimo de todos.
Palabras clave: angioedema, angioedema hereditario, C1 inhibidor, acceso
Received: 23-IX-2016 Accepted: 22-V-2017
Postal address: Eloisa Malbrán, Av. Roque Sáenz Peña 1160 1ro. “B”, 1035 Buenos Aires, Argentina
e-mail: elomalbran@hotmail.com
Hereditary angioedema (HAE) caused by deficiency of the C1 inhibitor (C1-INH-HAE) is a rare, autosomal dominant disease characterized by recurrent angioedema attacks of the skin and mucous membranes. It affects 1/50 000 of the population, with no race or sex difference and has an asphyxia related death rate of 15 to 50%1, 2. In the last 10 years, several new drugs for HAE treatment have been introduced into the market to better prevent and treat disease attacks. All new treatments proved efficacious in double blind, placebo controlled and prospective studies and are approved for its use across the world3. In Argentina, however, access to treatment is neither uniform nor universal.
Health delivery is a multiple steps process that begins when a patient recognizes that he is in need of a health service and ends with a measurable outcome of the subject. In Argentina, access to treatment is not yet universal and presents tremendous differences among our patient population. Those with lower socioeconomic backgrounds have usually poorer health outcomes, showing that equity still poses a huge challenge for some groups in society.
In our previous description of a cohort of 58 patients with hereditary angioedema, the mean delay to diagnosis was 15.3 years and the historical asphyxia related mortality was 13% (19 out of 148 individuals)5. Plasma derived C1 INH had been used at least once by 25% of subjects, but recurrently by scarcely 3%. We became aware that early recognition of the disease was crucial to avoid unnecessary HAE induced morbidity and mortality. The Argentina HAE patient association began a sustained campaign to identify and register all HAE patients and to educate physicians about HAE. Shire HGT introduced icatibant (Fyrazir@) into the country in 2010, and, together with CSL Behring for its plasma derived C1 INH concentrate (Berinert@), both began a marketing campaign that helped to disseminate the knowledge on HAE in the medical community. The lack of a national guide for HAE treatment was recognized as an important barrier to access; therefore an Argentina´s HAE treatment guide was published in 2012.
Lack of full coverage proved to be one of the main access barriers. In 2014, the Ministry of Health included pdC1-INH in a reimbursement program as a medication for low prevalence with high economic impact diseases8. Although this regulation does not facilitate access to all patients, it is supposed to guarantee supply to a great number of them. As a consequence, a new study to assess access to treatment was required to evaluate the impact of the new reimbursement rule and to identify new barriers. In this work we expanded our observations to at least 156 patients.
Materials and methods
The Argentina HAE Patient Association (AHAEPA) registers all patients that have a diagnosis of HAE confirmed by an HAE expert physician and have a typical disease history. If the diagnosis of HAE is in doubt, the patient is not entered into the registry.
The present work is an observational study on a disease registry that evaluates the health system of Argentina, and, as such, is exempted of ethical revision according to Resolution 1480-2011, scope and 5b, Argentina Ministry of Health. This was accepted and confirmed by our Bioethics Committee, Research Network.
A web based SurveyMonkey questionnaire was prepared by the authors and sent to 225 members of the AHAEPA by the responsible of the organization’s database. Answering to each question was elective for the members. A brief introductory note explaining the objectives of the survey was included. All data were obtained by the patient association and the identity of the participants was held confidential to the rest of the authors.
The following questions were submitted: What drug (if any) do you have available to treat an HAE acute attack? How much reimbursement do you get? How fast is the replacement process of used doses? Do all affected members in your family group have access to HAE medications? How early do you treat your symptoms? Do you self-inject?
The rationale behind these questions is that an acute moderate to severe HAE attack should be treated early enough to guarantee appropriate symptom control. If the treatment is delayed, it becomes useless, due to the short activation phase of bradykinin production followed by the spontaneous resolution of the episode9. Lack of reimbursement, replacement delays, sharing of medication among several members and inability to self-treat, all result in delays and a poor management of the condition. Additionally, some patients may still not be convinced of the benefits of early treatment.
All data was analyzed using the software of the survey program.
Results
The number of HAE patients in the registry increases constantly. By March 2016, 412 patients were included, representing approximately 50% of all theoretical cases, assuming a prevalence of 1/50 000 of the general population. Two hundred and twenty five emails were sent. Three of them were returned as email failure. One hundred and sixteen answers were obtained after the first email. A second and a third set of emails were sent, with a total of 156 (70%) of the questionnaires finally answered. Not all questions were answered in all occasions.
Medication availability, in comparison with our previous studies, is summarized in Table 1. The percentage of patients with available treatment for the next acute HAE attack has remained relatively constant through time even though the number of observations has almost tripled. It is interesting to note that these questionnaires are representatives of more than an individual each, since many answered on behalf of their kids or older relatives. It is to note that there has been a significant increase in the use of Firazyr since our last survey.
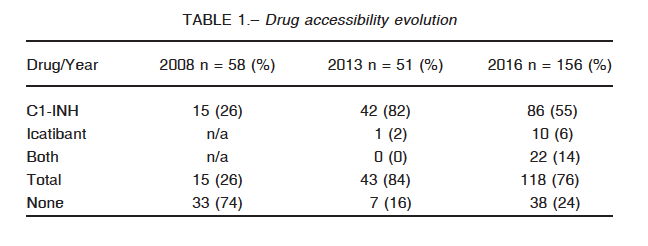
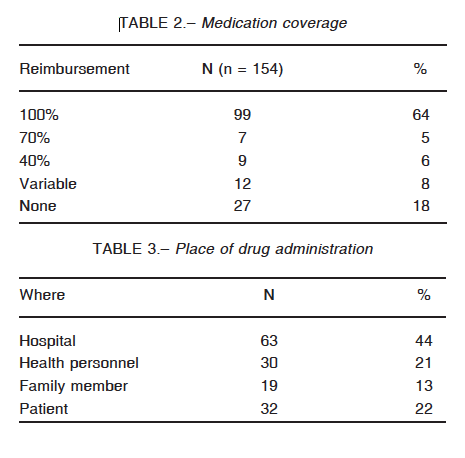
Several barriers to accessibility were identified. In the first place, coverage is not universal: only 63% of the patients have full reimbursement of the medication. Nineteen percent of them do not have any coverage at all and the rest has different discount percentages (Table 2). Next, replacement of medication presents enormous difficulties with delays going from 7 to 10 days, as reported by 45 (38%) of the participants. Forty eight subjects (40%) referred to this process as being exceedingly long and strenuous. Only 27 patients (23%) have fast access to new doses. There is no clear explanation for the delay. Most of the patients have to go through a complicated and annoying paperwork procedure. For some of them, a new set of laboratory proof of their C1 INH levels is required, every few months, irrespective of the genetic nature of this disease. Unjustified audits of the case records contribute to the delays.
Forty-three patients (35%, n = 124) answered that only one or a few members of the family group have access to the health system, and they share their medication on a first come first serve basis. Many patients are out of the legal workforce; they have neither health insurance nor access to reimbursement and cannot afford the price of medications. Others belong to health programs that are excluded of the Ministry of Health reimbursement list and receive variable drug price discounts or none, in any case, not enough to guarantee sustained access to treatment.
As a consequence of difficulties in reimbursement, delays in replacement treatments and medication sharing among family members, patients postpone treatment. Seventy-one patients (49%, n = 142) choose to receive medication at the beginning of the attack, while 73 (51%) wait until they experience severe pain or discomfort.
Finally, even though an effort has been made to establish the idea of early self-treatment, most patients seek medical or family assistance to receive the medication (Table 3).
Discussion
Hereditary angioedema due to C1 inhibitor deficiency is a rare disease that has a major impact on quality of life and asphyxia related mortality ranges from 15 to 50%. In the past, patients relied only on impeded androgens as prophylactic treatment and on fresh frozen plasma to stop acute attacks. In only few countries in the world, Argentina among them, a plasma derived C1 inhibitor concentrate was available to treat an acute angioedema attack. With the recognition that bradykinin as the main mediator of angioedema, new drugs classes and better preparations of plasma derived and transgenic forms of C1-INH concentrates became worldwide available3, 11. However, access to these medications presents huge inequalities across the world. Furthermore, since universal eligibility and removal of financial barriers to health care conform the basis of a fair health system, accountability of access to high quality, prompt and accessible system is mandatory4.
In our description of a cohort of HAE patients, we found that all deaths occurred before diagnosis of HAE was established5. We ventured that after diagnosis was confirmed, patients would be more active in seeking appropriate medical care, not relying in the usual allergy based antihistamine and steroid treatments but struggling for adequate, effective management of their disease. A striking finding in this cohort was the fact that although a plasma derived C1-INH concentrate wase approved in the country in the early 1980’s, only 25% of the patients had ever used it and only 3% used it in every eligible attack.
Since the Argentina HAE patient association started its activities in 2004, their members have committed to the identification of patients and appropriate treatment. During all these years, they pursued a sustained effort to identify all HAE Argentina’s patients and managed to establish a registry of them through a solid entry mesh that has included 412 subjects, 50% of the theoretical prevalence number, assuming a prevalence of 1: 50 000 of the population. We speculate that a further 10% is diagnosed in the country but that those subjects have not approached the AHAEPA yet, probably accounting together for 60% of all country’s cases.
Several barriers to medication access in Argentina were progressively identified. Initially, health insurance audits rejected treatment due to the lack of a country guide to therapy. Consequently, an HAE treatment guide for Argentina was published in Medicina (B Aires) 6. This simple step partially contributed to improve access to treatment to 86% of our cohort subjects. However, new and unexpected access barriers to therapy were identified, being lack of universal coverage a major impediment to treatment.
A petition was made to the Ministry of Health of Argentina to include an HAE treatment for acute attacks within the country’s program for rare diseases with high economic
impact. This was accomplished in 2014. After 15 months of extended coverage, we conducted the present study to evaluate the impact of the new reimbursement policy, to identify new possible access to treatment and extend our observations to HAE patients beyond our cohort. In this way, a better statistical sample, more representative of the real situation in our country, would be obtained. Also, we meant to study the impact of AHAEPA sponsored patient meetings and self-infusions clinics on promotion of early treatment for the successful control of the acute attack and management of the condition.
We used a web based approach to contact the patients. In order to maintain the confidentiality of the data, personal identifiers were held occult to the authors that do not pertain to the AHAEPA. Most patients in the database with a known email address, almost 50%, were approached and roughly 70% of answers were collected representing at least 18% of the total theoretical number of patients. This number, however, is probably an underestimation, since the participant answering the survey may also have kids with HAE or older relatives who did not actively participate in our study.
Our results identify several hidden barriers to access to medication in this population10. The first one continues to be lack of universal coverage. Although 80% of the patients have access to drugs, some have them through other family members. Informal jobs without health insurance or formal jobs not included in minister of Health reimbursement ruling were the main causes. There is space to improve access using different tools like local government support or public hospital budgeting. The health medical organization´s inappropriate and unjustified operational delays to drug delivery and replacement needs to be addressed through quality assurance programs and state ruling. Taking into account the availability of HAE treatments in our country, there is no justification to patient suffering and constantly claiming for access to these life saving therapies. The current access barriers lead all affected members within a family to share medications and to delay the timing of treatment administration to preserve the dose for a hypothetical more severe attack that might happen. Finally, even though a consistent effort to train patients has been made, the goal of early self-injection is, yet, out of the reach of most of our patients11.
Conflict of interests: None to declare.
References
1. Frank MM, Gelfand JA, Atkinson JP. Hereditary angioedema: the clinical syndrome and its management. Ann Intern Med 1976; 84: 589-93.
2. Agostoni A, Cicardi M. Hereditary and acquired C-1 inhibitor deficiency: biological and clinical characteristics in 235 patients. Medicine (Baltimore) 1992; 71: 206-15.
3. Cicardi M, Bork K, Caballero T, et al. on behalf of HAWK (Hereditary Angioedema International Working Group). Evidence-based recommendations for the therapeutic management of angioedema owing to hereditary C1 inhibitor deficiency: consensus report of an International Working Group. Allergy 2012; 67: 147-57.
4. Solberg L, Mosser G, McDonald S. The three faces of performance measurement: improvement, accountability, and research. Jt Comm J Qual Improv 1997; 23: 135-47.
5. Fernández Romero DS, Di Marco P, Malbran A. Hereditary angioedema. Family history and clinical manifestations in 58 patients. Medicina (B Aires) 2009; 69: 601-6.
6. Malbrán A, Fernández Romero DS, Menéndez A. Hereditary angioedema. A therapeutic guide. Medicina (B Aires) 2012; 72: 119-23.
7. Malbrán A, Malbrán E, Menéndez A, Fernández Romero DS. Hereditary angioedema. Treatment of acute attacks in Argentina. Medicina (B Aires) 2014; 74: 198-200.
8. Resolución 1048/14. Superintendencia de Seguros de Salud. Ministerio de Salud. República Argentina. In: http://servicios.infoleg.gob.ar/infolegInternet/anexos/230000-234999/231401/texact.htm; accesed on 09/15/2016.
9. Bork K, Staubach P, Eckardt AJ, Hardt J. Symptoms, course, and complications of abdominal attacks in hereditary angioedema due to C1 inhibitor deficiency. Am J Gastroenterol 2006; 101: 619-27.
10. Gulliford M, Figueroa-Munoz J, Morgan M, et al. What does ‘access to health care’ mean? J Health Serv Res Policy 2002; 7: 186-8.
11. Zuraw BL, Banerji A, Bernstein JA, et al. US Hereditary Angioedema Association Medical Advisory Board 2013 recommendations for the management of hereditary angioedema due to C1 inhibitor deficiency. J Allergy Clin Immunol Pract 2013; 1: 458-67.
– – – –
IMPACTANTE
“¿Que me dice de la Moncha?”. “¡Impactante!”. (Eisenstein, Tratado de álgebra.)
Adolfo Bioy Casares (1914-1999)
Breve diccionario del argentino exquisito. Buenos Aires: Emecé, 1978, p 79